INTRODUCTION
Glutathione (GSH) is a low molecular weight tripeptide (gamma-glutamylcysteinelglycine) which is synthesized intracellularly. It plays a critical role in the detoxification of several drugs and xenobiotics (Meister and Anderson, 1983) and in cellular defence against agents that cause oxidative stress (Anderson, 1998). The protective action of GSH is based on its ability to oxidize the thiol group of its cysteine, which leads to the formation of oxidized glutathione (GSSG). In the glutathione cycle, GSH is regenerated via the antioxidant enzyme glutathione reductase as GSSG reacts with NADPH+H+ resulting in the formation of 2GSH molecules+NADP+. This reaction is irreversible and accounts for the high ratios of GSH: GSSG found in the cells (Miester and Anderson, 1983).
A very well established technique to measure GSH in cultured cells is to add the cell permeant, fluorogenic vital dye monochlorobimane (MCB) to the growth medium to detect both GSH levels and activity in cells. MCB does not fluoresce, but on reacting with GSH yields GSH-bimane adducts which fluoresce. The enzyme glutathione-S-transferase (GST) exclusively mediates the intracellular conjugation of GSH and MCB. The rate of conjugation between GSH and MCB (which produces the fluorescence signal) is dependent on the abundance of GST (Haugland, 2005).
GSH and GST studies have been performed on parasites as a functional dissection of the parasite's oxidative defence system, which may improve chemotherapy. Ribeiro et al. (1998) demonstrated a decrease in GSH levels in S. mansoni exposed in vitro to the anti-schistosomal drug, praziquantel and suggested that GSH depletion in schistosomula might render them susceptible to the host immune response. The importance of thiol metabolism to trypanosome survival has been suggested in several studies. Trypanosoma spp. contain glutathione but lack glutathione reductase activity. Instead, the parasite has evolved an analogous system with a novel cofactor trypanothione (TSH), which consists of a spermidine moiety linked to 2 GSH molecules (Fairlamb et al. 1985). Investigation of the oxidative defence system of T. cruzi has revealed that this parasite expresses cytosolic, mitochondrial (Wilkinson et al. 2000) and glycosomal peroxidases (Wilkinson et al. 2002) whose activities are linked to the reduction of the parasite-specific thiol trypathione. TSH is the major reduced thiol in Leishmania (Fairlamb and Cerami, 1992) and GSH (detected using MCB) in Toxoplasma gondii (Stommel et al. 2001). The importance of thiol metabolism to Cryptosporidium survival has not been investigated, but Entrala et al. (1997) detected an NADPH-dependent H2O2 scavenging system in insoluble fractions of C. parvum oocysts using biochemical assays.
Treatment of cells with sulfoximine inhibitors of γ-glutamylcysteine synthetase is an excellent method for depleting intracellular GSH. Buthionine sulfoximine (BSO) is a potent and selective inhibitor of GSH synthesis and is highly effective in vivo and in vitro. Injection of mice with BSO produces a rapid decrease in GSH levels in plasma, kidney, liver and other tissues (Miester and Anderson, 1983). Ribeiro et al. (1998) demonstrated that exposure to BSO in vitro caused a decrease in S. mansoni schistosomular GSH levels. Mammalian cells depleted of GSH by BSO showed increased endogenous oxidative damage (Will et al. 1999).
Ionizing radiation (UV light and gamma irradiation) induces the production of reactive oxygen species (ROS) in cells, which can damage cellular elements. Oxidative modifications to DNA nucleotides (e.g. 8-hydroxyguanine; 8-oxoG) are mutagenic (Cheng et al. 1992). Antioxidants reduce ROS production and GSH plays a significant role in inhibiting the generation of 8-oxoG by ionizing radiation (Fischer-Nielsen et al. 1994). UV irradiation ([ges ]4 mJ.cm−2) alters the DNA structure of C. parvum oocysts and renders them non-infectious to susceptible neonatal mice (Rochelle et al. 2005).
Here, we treated C. parvum oocysts with the fluorogenic vital dye MCB to assess the following: (i) localization of GSH in C. parvum oocysts and sporozoites, (ii) the effect of buthionine sulfoximine (BSO) on MCB localization in sporozoites (iii) oocyst viability and (iv) its usefulness for determining whether UV irradiation damage affects sporozoite GSH levels in intact oocysts.
MATERIALS AND METHODS
Parasite
Purified C. parvum oocysts (Iowa isolate) were purchased from Bunch Grass Farm (BGF, USA) and stored between 4 and 8 °C until used. Viability was determined by both the fluorogenic vital dyes assay of Campbell et al. (1992) and the maximized in vitro excystation assay of Robertson et al. (1993).
Labelling of oocysts
Viability of oocysts was determined using monochlorobimane and propidium iodide and compared with the fluorogenic vital dye assay.
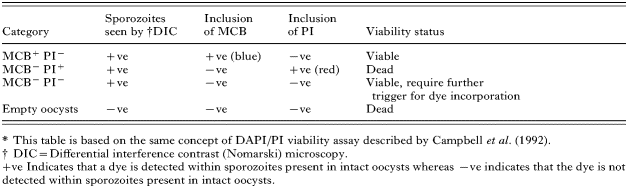
Working solutions of 10 mM MCB (2·26 mg ml−1 in ethanol) and 1·5 mM propidium iodide PI (1 mg ml−1 in PBS) were prepared and kept on ice. Oocysts (1×106) were acidified in 1 ml of Hanks' balanced salt solution (HBSS), pH 2·75, then incubated for 1 h at 37 °C. Oocysts were washed 3 times with HBSS (pH 7·2), re-suspended in 100 μl of HBSS, then incubated simultaneously with 10 μl of MCB working solution (1 mM) and 10 μl of PI working solution (0·15 mM) for 3·5 h at 37 °C. Following incubation, 5 μl of FITC-labelled-anti-Cryptosporidium monoclonal antibody (FITC-CmAb, ×20, Waterborne Inc, USA) were added and samples were incubated for a further 30 min. Labelled oocysts were washed 3 times with HBSS and analysed immediately by epifluorescence microscopy. Control oocysts were labelled with 4,6-diamidino-2-phenylindole (DAPI) and PI following the protocol of the fluorogenic vital dye assay as described by Campbell et al. (1992). Ten μl samples of labelled oocysts were viewed under both Nomarski differential interference contrast (DIC) and epifluorescence microscopy. A total of 100 oocysts were enumerated on 3 separate occasions for each sample and categorized as follows: MCB positive/PI negative (MCB+ PI−), MCB negative/PI positive (MCB− PI+), MCB negative/PI negative (MCB− PI−) and empty oocysts (Table 1).
Oocyst excystation
We followed the maximized in vitro excystation method of Robertson et al. (1993). Stock solutions of bile (1% bovine bile in Hanks minimum essential medium, HMEM) and sodium hydrogen carbonate (0·4% NaHCO3 in distilled water) were prepared. Oocysts (1×106 ml−1) were acidified in 1 ml of HBSS (pH 2·75) for 1 h at 37 °C. Following acidification, oocysts were washed thoroughly and re-suspended in 100 μl of HBSS (pH 7·2). Ten μl samples of acidified oocysts were placed on slides and oocysts and sporozoites enumerated under DIC optics. Oocysts suspended in 100 μl of HBSS were treated with 250 μl of a freshly prepared mixture of 200 μl bile and 50 μl NaHCO3 and incubated for 30 min at 37 °C. Following incubation, 10 μl samples of oocyst suspension were removed and examined for excysted sporozoites, partially excysted oocysts and empty oocysts. Oocysts were incubated for a further 3·5 h at 37 °C and following this the proportions of intact, partially excysted and empty oocysts were determined.
Inhibition of glutathione synthesis in oocysts
To determine whether GSH synthesis occurs in sporozoites within intact oocysts, oocysts were treated with buthionine sulfoximine (BSO), a specific GSH synthesis inhibitor. Samples (100 μl) of pre-acidified oocysts were incubated with different concentrations of BSO (1, 2·5 and 5 mM) for 4 h at 37 °C, then thoroughly washed and labelled with MCB, PI and FITC-CmAb as described above. Two control groups were labelled with MCB/PI or DAPI/PI without prior BSO treatment. Both MCB and/or PI and DAPI and/or PI oocysts (Table 1) were enumerated and the fluorescence intensity of individual oocysts was determined by fluorescence quantification using the Analysis™ system (Olympus, UK).
UV irradiation of oocysts
C. parvum oocysts were exposed to a low-pressure UV lamp with an output at 254 nm. The procedure was adapted from the method described by Rochelle et al. (2004). Short-wave UV irradiation from a UVGL-58 Mineralight lamp was used. The intensity of the UV light, measured using a digital UVX radiometer, was (on average) 350 μW.cm−2 at 254 nm. A rig was set up 10 cm below the lamp, and a position marked where the UV intensity was maximal (350 μW.cm−2). The UV dose was then determined from: UV dose=Irradiance×Exposure time (sec), mJ.cm−2=mW.cm−2×sec.
In all experiments, 1×106 oocysts were suspended in 5 ml of HBSS. Samples were placed in Petri dishes (36 mm diameter) which were constantly mixed using a magnetic stirrer during exposure to UV light. To achieve different UV dosages (mJ.cm−2), oocysts were exposed to UV light for varying times at a constant distance (10 cm) from the constant intensity UV source. For each experiment, control oocysts were kept under the same conditions without UV irradiation.
Preparation of sporozoites
Approximately 1×106 C. parvum oocysts were excysted as described above. Freshly excysted sporozoites were divided into 2 groups. In the first group, sporozoites were fixed in suspension in 5% methanol in HMEM for 5 min then washed 3 times in HBSS by centrifugation for 10 sec at 14000 g (Beckman, UK). Fixed sporozoites were purified through a 3 μm cellulose acetate membrane filter (Millipore, UK) which entrapped empty oocysts and other large particulates. Five μl of filtrate, containing purified sporozoites, were pipetted individually onto 30-well multispot microscope slides (Hendely-Essex, UK) and left to air dry at room temperature. In the second group, primary fixation in 5% methanol was omitted, and unfixed, purified sporozoites were air dried at room temperature onto slides before being fixed in methanol. Sporozoites in both groups were treated with 0·2% Triton-X-100 (Sigma) in HBSS for 3 min to increase surface membrane permeability. Slides were washed thoroughly in HBSS, air dried then labelled with MCB. Slides were incubated with serial dilutions of MCB in HBSS (1[ratio ]2 to 1[ratio ]1024) for 3 h at 37 °C in a humidified chamber. Following incubation, slides were washed 3 times in HBSS and analysed by both DIC and epifluorescence microscopy (see below).
Microscopy
Microscopy was performed on an Olympus BH2 microscope equipped with DIC and epifluorescence optics, using the following filter sets: 450 nm – emission/350 nm – excitation (sky blue) for MCB and DAPI and 630 nm – emission/500 nm – excitation (red) for PI. Photography was performed using a ColorView (Soft Imaging Systems Inc.) digital camera attached to the Olympus BH2 microscope. In some experiments the fluorescence of labelled oocysts was quantified using the Analysis™ system (software for scientific imaging and calibrated image measurements; Olympus, UK). All oocyst enumerations were performed at either ×500 or ×1250 total magnification and the localization of all fluorogens and organelles was investigated at ×1250 total magnification. Images shown represent at least 3 experiments.
Statistical analysis
Statistical analysis was performed using the analysis of variance test (ANOVA) with P<0·05 as the criterion of significance using the MINITAB version 11 programme.
RESULTS
Monochlorobimane uptake by C. parvum oocysts and sporozoites
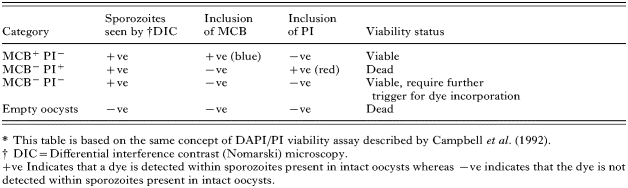
Three factors were considered in determining the optimum labelling of pre-acidified oocysts with MCB: MCB concentration, temperature and incubation time. Oocysts were incubated in 3 different MCB concentrations (0·1, 0·5 and 1 mM). A time-course for maximizing MCB uptake was determined, whereby oocysts were incubated with MCB for 1, 2 and 4 h at 37 °C. A significant uptake of MCB occurred after 4 h incubation with an MCB concentration of 1 mM (Fig. 1). Therefore, in all subsequent experiments oocysts were incubated with 1 mM MCB for 4 h at 37 °C.
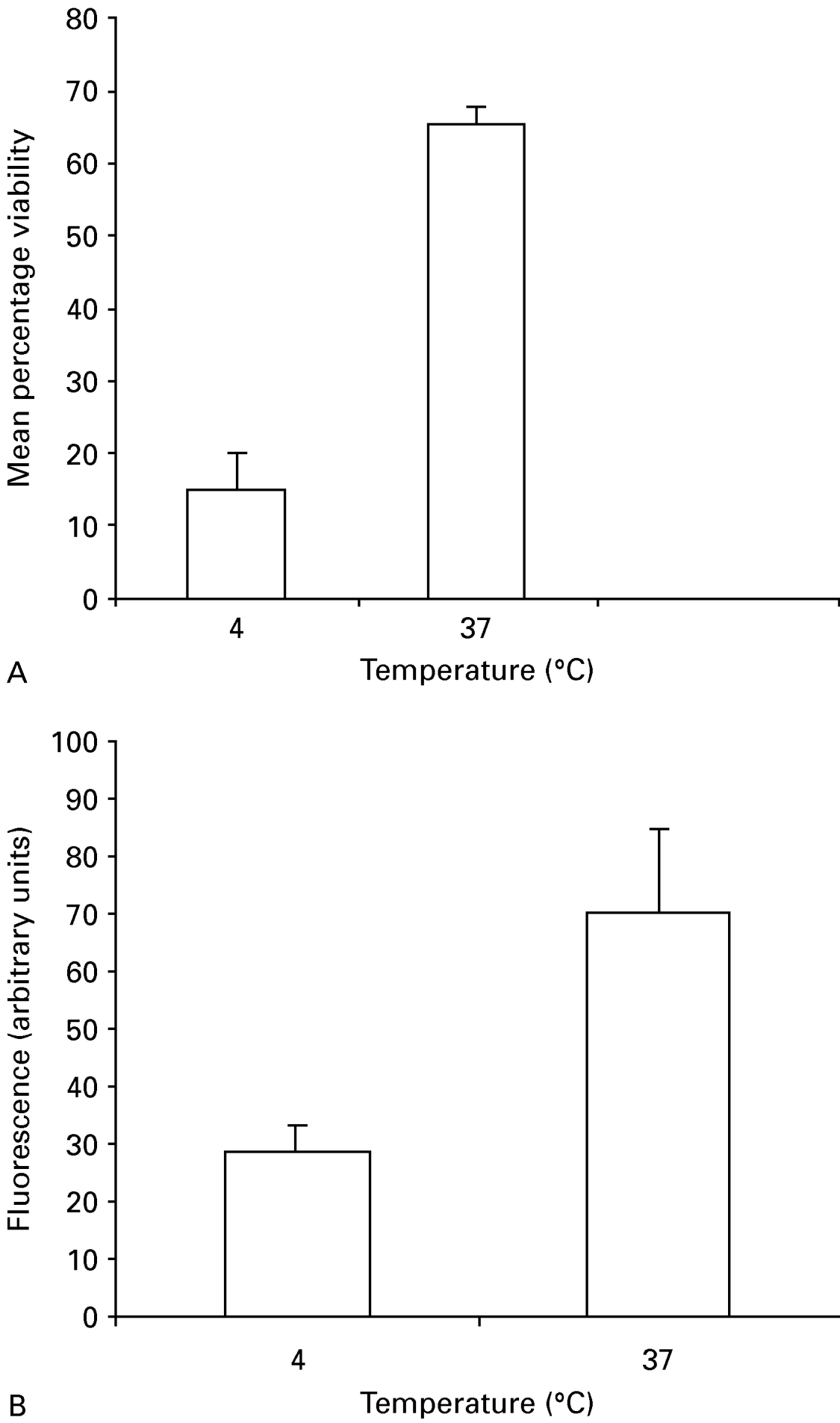
Fig. 1. Effect of temperature on MCB uptake by Cryptosporidium parvum oocysts. Oocysts were incubated with MCB/PI and FITC-CmAb at 4 °C or 37 °C for 4 h. After incubation, oocysts were washed and enumerated as described for the MCB/PI viability assay (A) and their fluorescence quantified (B). Histograms represent mean percentage of viable oocysts (A) and mean fluorescence/oocyst (B). Error bars present standard deviations, (n=300) in (A) and (n=100) in (B).
MCB fluorescence was used to localize GSH in intact oocysts (Fig. 2). The number of distinctly labelled foci in intact oocysts varied between 2 and 6 (Fig. 2A, B and E) in recently excreted (10 days old) and aged (6 months old; data not shown) oocysts labelled with MCB. We investigated MCB localization in freshly excysted, purified, methanol-fixed sporozoites. MCB-labelled sporozoites retained the fluorogenic dye at several intra-sporozoite foci (Fig. 2C and D). GSH distribution in sporozoites appeared to be granular in the apical and posterior (nuclear) regions. A similar labelling pattern was obtained when sporozoites were air dried onto slides prior to fixation, but the fluorescence intensity was reduced.

Fig. 2. Cryptosporidium parvum oocysts simultaneously labelled with MCB/PI (A–B and E–G) or DAPI/PI (H–J). Viable oocysts stain with MCB or DAPI and dead oocysts stain with PI. DAPI or MCB-labelled oocysts fluoresce blue (left panel) and PI-labelled oocysts fluoresce red (middle panel). The right panel contains the same images viewed under Nomarski differential interference contrast (DIC) optics. (C–D) Localization of MCB in methanol-fixed sporozoites. C. parvum sporozoites were fixed in 5% methanol, washed, purified and labelled with MCB for 3 h at 37 °C. The middle panel represents MCB-labelled sporozoites whereas the right panel represents the same image viewed under DIC. n, nucleus. Arrows point to the intra-sporozoite localization of MCB in both intact oocysts (A, B and E) and purified sporozoites (C and D).
Determinination of oocyst viability using MCB and PI staining
The procedure developed was similar to that used in the fluorogenic vital dyes (DAPI/PI) assay of Campbell et al. (1992) and the results are shown in Figs 2 and 3. The proportions of MCB+ PI−, MCB− PI+, MCB− PI− and empty oocysts were quantified by enumerating 100 oocysts in triplicate samples. Oocysts were considered viable if they did not include PI but were stained with MCB (MCB+ PI−). Also, intact oocysts which did not include MCB or PI (MCB− PI−) but contained sporozoites under DIC were considered viable (Table 1).

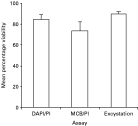
Fig. 3. Viability of Cryptosporidium parvum oocysts (Iowa isolate) determined by 3 different methods: DAPI/PI, MCB/PI vital dyes assays and maximized in vitro excystation assay. Histograms represent mean percentage viability of 3 different experiments. Error bars are standard deviations (n=300) per experiment.
MCB incorporation correlated well with DAPI+PI− oocyst staining (Fig. 2 E–G and H–J). The results of DAPI/PI and MCB/PI staining were not significantly different when labelled oocysts were enumerated (mean±S.D. 84·7±4·2 vs 73·3±8·7, P=0·05) respectively. Results using the maximized in vitro excystation assay were not significantly different from DAPI/PI staining (mean±S.D. 89·7±2·1 vs 84·7±4·2, P=0·1) but were significantly different from MCB/PI staining (mean±S.D. 89·7±2·1 vs 73·3±8·7, P=0·01).
Glutathione depletion inhibits binding of monochlorobimane in oocysts
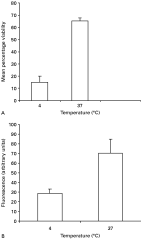
We used buthionine sulfoximine (BSO), a potent and specific inhibitor of GSH (Meister and Anderson, 1983), to determine whether GSH is synthesized in BSO-treated oocysts, by labelling treated oocysts with MCB. Three different BSO concentrations were tested (1, 2·5 and 5 mM). The percentage inhibition was determined by enumerating 100 oocysts in 3 consecutive experiments. A significant inhibition of MCB fluorescence (reflecting MCB uptake) was observed in GSH depleted oocysts with 5 mM BSO (mean±S.D. 35±3·7) compared with the control group (3·3±1·2, P=0) (Fig. 4A). BSO-treated oocysts showed a clear reduction in fluorescence within viable oocysts but no change in the labelling pattern was observed in PI-positive (dead) oocysts. Incubation of oocysts with 5 mM BSO for 4 h at 37 °C induced spontaneous excystation and increased the number of empty oocysts by about 30%. To minimize oocyst excystation and the production of empty oocysts, all further incubations with BSO were conducted using 1 mM BSO for 24 h at room temperature. 1 mM BSO-treated oocysts exhibited weak or no MCB fluorescence, although they were not PI positive (dead), but were intact and contained sporozoites by DIC. As shown in Fig. 4B, significant inhibition of MCB fluorescence was observed in BSO-treated oocysts (mean±S.D. 50·6±9·7) compared to the untreated group (15·5±9·6, P=0). Similar results were obtained when the fluorescence of BSO-treated oocysts was quantified. Oocysts subjected to glutathione depletion showed significant decrease in GSH-bimane adducts (mean±S.D. 62·3±13·4) when compared to the control group (mean±S.D. 165·1±20·3, P=0) (Fig. 4C).
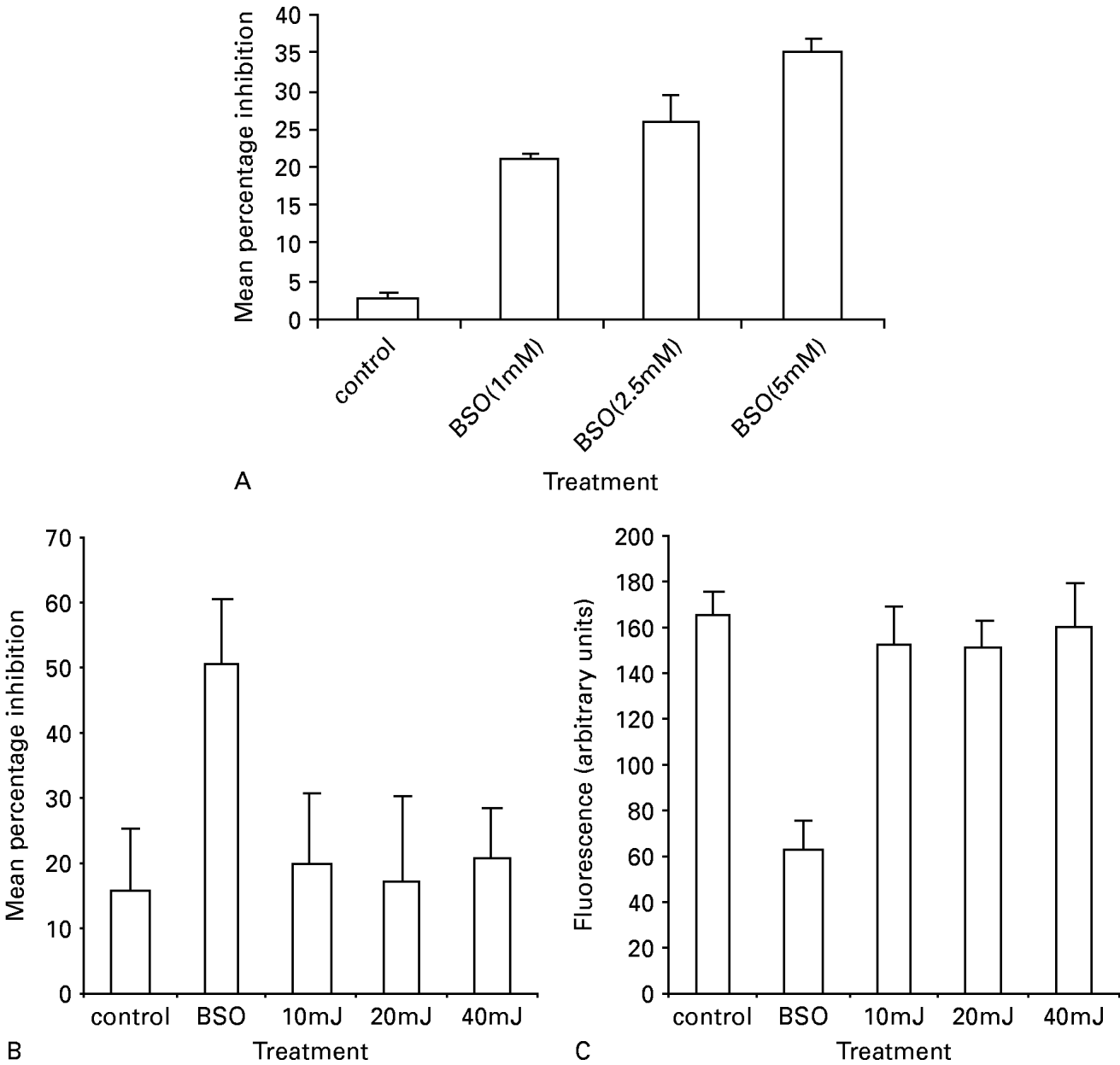
Fig. 4. (A) Effect of different concentrations of BSO on MCB uptake by Cryptosporidium parvum oocysts. Oocysts were incubated for 4 h at 37 °C with 1, 2·5 and 5 mM BSO. Following incubation, oocysts were washed and labelled with MCB/PI and FITC-CmAb for 4 h at 37 °C. BSO-treated oocysts exhibited weak or no MCB fluorescence, although they were not PI positive (dead), but were intact and contained sporozoites by DIC. Histograms represent mean±S.D. (n=100). (BandC) Effect of BSO treatment (1 mM for 24 h at room temperature) or UV irradiation (10, 20 or 40 mJ.cm−2 doses of UV light) on MCB uptake by C. parvum oocysts. Oocysts were treated as described in (A) except for the incubation conditions with BSO and enumerated (B), and their fluorescence quantified (C). Histograms in (B) represent percentage inhibition±S.D. (n=300) and histograms in (C) represent oocysts fluorescence intensity (arbitrary units)±S.D. (n=100).
Effect of UV irradiation on monochlorobimane uptake by C. parvum oocysts
Oocysts were exposed to 10, 20 or 40 mJ.cm−2 doses of UV light, enumerated for MCB inclusion or exclusion, and their fluorescence quantified. The percentage inhibition of irradiated versus control oocysts was not significantly different (irradiated group mean±S.D. 19·8±10·5, 17·2±12·8, 20·6±8·0 at 10, 20 or 40 mJ.cm−2 respectively; control group mean±S.D. 15·5±9·6, P>0·05). No significant differences in fluorescence intensity or distribution of MCB occurred in irradiated or control oocysts (irradiated group mean±S.D: 10 mJ.cm−2=152·3±15·9, 20 mJ.cm−2=149·6±12·2, 40 mJ.cm−2=160·2±18·5; control group mean±S.D. 165·1±20·3, P>0·05) (Fig. 4C).
DISCUSSION
In the present study, evidence for GSH localization in C. parvum sporozoites was inferred from experiments in which intact oocysts were stained with molecular probes and antibodies. Using the GSH-sensitive fluorogenic dye, MCB and FITC-CmAb, we demonstrated that MCB localized at several distinct intra-sporozoite foci including the nucleus and cytoplasm. Bellomo et al. (1992) demonstrated nuclear compartmentalization of glutathione using intact mammalian cells labelled with MCB in vitro. Furthermore, the nuclear/cytoplasmic GSH concentration gradient was maintained by an active cellular mechanism. This finding was confirmed by Briviba et al. (1993) who detected the subcellular distribution of MCB in rat hepatocytes following its microinjection into their cytoplasm. Fluorescent accumulations were observed in the nucleus within 1–2 sec.
The importance of thiol metabolism in survival has been investigated in protozoan parasites such as Trypanosoma (Fairlamb et al. 1985; Wilkinson et al. 2000; Wilkinson et al. 2002), Leishmania (Fairlamb and Cerami, 1992) and Toxoplasma (Stommel et al. 2001), yet evidence for glutathione as a Cryptosporidium survival mechanism is minimal. Using biochemical assays, Entrala et al. (1997) detected low levels of superoxide dismutase and an NADPH-dependent-H2O2 scavenging system in the insoluble fractions of C. parvum oocysts. Our data, demonstrating the presence of nuclear and cytoplasmic foci staining with MCB in both intact, viable oocysts and purified sporozoites support the presence of GSH in C. parvum. In addition, we identified C. parvum homologues of the human GST (NP_384895.1) and the glutathione peroxidase (cgd3-460, EC 1.11.1.9) genes following searching the ‘crypto genome project’ database, which further argues for the presence of GSH and GST in C. parvum.
MCB can be used to detect glutathione in C. parvum oocysts. However, its use alone is insufficient to determine their viability. The fluorogenic vital dye assay of Campbell et al. (1992) is an effective and reliable method to determine C. parvum oocyst viability. Thus, we anticipated that adopting the same protocol for MCB/PI staining would be as effective. This included pre-acidification of oocysts prior to the addition of vital dyes. Our prediction of differential coloration of the nuclei of MCB (+ve)/PI (−ve) (viable) oocysts (which stain blue) and MCB (−ve)/PI (+ve) (dead) oocysts (which stain red) was confirmed by epifluorescence microscopy and supported by DIC examination. Using a combination of MCB, PI and FITC-CmAb enabled discrimination between viable and non-viable oocysts, indicating that MCB and PI might be a useful viability assay. While the prediction of oocyst viability using the MCB/PI assay was comparable to that of the DAPI/PI assay (mean±S.D. 73·3±8·7 vs 84·7±4·2, P=0·05), its prediction differed from the maximized in vitro excystation assay (mean±S.D. 73·3±8·7 vs 89·7±2·1 P=0·01). This discrepancy could be due to differences in identifying and enumerating oocysts containing readily recognizable DAPI positive nuclei as compared to the more variable, more diffuse staining seen in MCB-stained oocysts.
We examined the effect of temperature and BSO treatment (a specific inhibitor of GSH) on the staining of oocysts with MCB/PI. MCB uptake and labelling of intact oocysts did not occur at 4 °C therefore, MCB uptake appears to be energy-dependent, dependent upon the metabolic activity of viable, unexcysted sporozoites. Oocysts depleted of their GSH following BSO treatment demonstrated ~50% reduction in MCB uptake, indicating that GSH synthesis occurs in sporozoites in intact oocysts. Our treatment conditions (incubation with 1 mM BSO for 24 h at room temperature) are probably not sufficiently lengthy to permit total GSH depletion in sporozoites. A similar finding has been reported in Schistosoma mansoni (Ribeiro et al. 1998). The MCB/PI assay can be used to determine the effects of temperature and chemical insults which affect the metabolic activity of sporozoites.
In cultured mammalian cells, irradiation (Mansur et al. 2001) and thermal stress (Will et al. 1999) induce DNA base modifications with the production of reactive oxygen species (ROS) such as peroxidase and hydroxyl radicals. GSH reacts with various ROS and is a cofactor for the H202-removing enzyme glutathione peroxidase. Depletion of GSH and thermal stress increase endogenous oxidative damage, but the addition of thiols to the medium does not reduce the level of oxidative damage (Will et al. 1999). The protective effects of reduced glutathione against UV-B-induced damage has been reported in the nitrogen-fixing cyanobacterium (Nostoc muscorum) (Tyagi et al. 2003). They suggested that the presence of reduced glutathione and certain other reducing agents in the natural habitat or within cells of living organisms may partially protect or repair the damaging effects of UV-B irradiation. UV irradiation ([ges ]4 mJ.cm−2) alters the DNA structure of C. parvum oocysts and renders them non-infectious to susceptible laboratory hosts although they are viable using in vitro surrogates (Rochelle et al. 2005). In our study, irradiation, at doses of 10–40 mJ.cm−2, did not affect the uptake of MCB by C. parvum oocysts.
Little is known about the mechanisms developed by protozoan parasites to cope with the generation of ROS. Brown et al. (1995) described a free radical detoxification mechanism, based on cysteine-rich proteins in Giardia duodenalis, which lacks glutathione. Entrala et al. (1997) could not demonstrate the presence of a glutathione-dependent enzyme system in C. parvum and suggested that oxidative protection might occur through the availability of high concentrations of free-radical scavengers like mannitol or alternative thiols. Sodium selenite (selenium) has an inhibitory effect on C. parvum infection in vitro and in vivo. Parasite numbers were significantly reduced in cell culture following treatment with selenium (Haung and Yang, 2002). Selenium-induced reduction in C. parvum infection was abrogated using a combined solution of free-radical scavengers of mannitol and reduced glutathione. Thus, selenium-induced oxidative stress is possibly a major mechanism in inhibiting C. parvum infection (Haung and Yang, 2002). Our data, based on the MCB-labelling of intact C. parvum oocysts, identify the presence of glutathione both in nuclear and cytoplasmic foci of sporozoites, which can be specifically depleted by BSO. Its function as an endogenous free radical scavenger in UV-irradiated oocysts was not demonstrated. Thus it is likely that other free radical scavengers are more active than GSH in UV treated C. parvum (e.g. cysteine, ascorbic acid). MCB is unlikely to be useful as a surrogate for detecting UV damage in UV-treated Cryptosporidium oocysts.
This work was funded by the Environmental and Rural Affairs Department, Agricultural and Biological Research Group, Scottish Executive, Scotland, UK. We thank Dr D. Reid, Drinking Water Quality Unit, Scottish Executive, 1-H(N) Victoria Quay, Edinburgh, EH6 6QQ for managing the project.