INTRODUCTION
Protozoan parasites of the genus Cryptosporidium continue to cause water-borne enteric disease throughout the world (Wheeler et al. Reference Wheeler, Vugia, Thomas, Beach, Carnes, Maier, Gorman, Xiao, Arrowood, Gilliss and Werner2007; Beaudeau et al. Reference Beaudeau, De Valk, Vaillant, Mannschott, Tillier, Mouly and Ledrans2008; Mason et al. Reference Mason, Chalmers, Carnicer-Pont and Casemore2010). Consequently, considerable effort has been invested in the development of in vitro culture techniques for a multitude of purposes. Such uses include the detection and enumeration of infectious oocysts from the environment (Slifko et al. Reference Slifko, Freidman, Rose and Jakubowski1997; LeChevallier et al. Reference LeChevallier, Di Giovanni, Clancy, Bukhari, Bukhari, Rosen, Sobrinho and Frey2003; Lalancette et al. Reference Lalancette, Di Giovanni and Prevost2010), quantification of oocyst inactivation from a variety of stresses responsible for attenuation of oocyst infectivity (Quintero-Betancourt et al. Reference Quintero-Betancourt, Gennaccaro, Scott and Rose2003; Li and Haas, Reference Li and Haas2004; Connelly et al. Reference Connelly, Wolyniak, Williamson and Jellison2007; Ives et al. Reference Ives, Kamarainen, John and Rose2007) and not the least, study of the parasite life cycle (Borowski et al. Reference Borowski, Thompson, Armstrong and Clode2010). However, in vitro cell culture has often been hampered by a number of factors, including low levels of infectivity affecting assay sensitivity (Rochelle et al. Reference Rochelle, Ferguson, Johnson and De Leon2001; Arrowood, Reference Arrowood2002; Schets et al. Reference Schets, Engels, During and Husman2005), as well as delayed life-cycle development and poor synchronicity affecting the precision and interpretation of data (Weir et al. Reference Weir, Pokorny, Carreno, Trevors and Lee2001).
Like other Apicomplexans, Cryptosporidium sporozoites contain a number of vesicular secretory organelles and apical organelle discharge has been identified as essential in sporozoite motility, secretion and infection (Chen et al. Reference Chen, O'Hara, Huang, Nelson, Lin, Zhu, Ward and Larusso2004; Huang et al. Reference Huang, Chen and Larusso2004). Significantly, recent work has demonstrated that once excysted, sporozoites rapidly undergo distinct physiological changes including apical organelle discharge of secretory organelles (Chen et al. Reference Chen, O'Hara, Huang, Nelson, Lin, Zhu, Ward and Larusso2004) accompanied by changes in membrane depolarization, increased sporozoite intracellular calcium and rapid reductions in internal energy resources (King et al. Reference King, Hoefel, Lim, Robinson and Monis2009; Matsubayashi et al. Reference Matsubayashi, Ando, Kimata, Nakagawa, Furuya, Tani and Sasai2010). The timing of this discharge is critical and needs to occur when the sporozoite is in close proximity or intimate contact with a compatible cell for successful infection to be established (King et al. Reference King, Hoefel, Lim, Robinson and Monis2009). While Cryptosporidium oocysts are long lived and resilient to numerous environmental and water treatment stresses (Smith et al. Reference Smith, Robertson and Ongerth1995), excysted sporozoites have a limited opportunity to infect a compatible cell (Upton et al. Reference Upton, Tilley, Nesterenko and Brillhart1994; Widmer et al. Reference Widmer, Klein and Bonilla2007). Importantly, oocysts may release their sporozoites before settling on a monolayer and in vitro assays have not necessarily been optimized bearing this in mind.
We therefore decided to investigate factors that may affect the timing of sporozoite contact with a target cell, in particular, to examine the relationship between excystation rate and the proximity of oocysts/excysted sporozoites to the cell monolayer and the effect on in vitro infectivity. We report that in a standard assay format the majority of sporozoites are not close enough to the cell monolayer to successfully establish infection. However this can be overcome by centrifugation of oocysts onto the monolayer, greatly increasing the sensitivity of the assay (Weir et al. Reference Weir, Pokorny, Carreno, Trevors and Lee2001). Secondly, excystation procedures can also be tailored to control the excystation rate, thereby influencing experimental design and data interpretation. Finally, we demonstrate that the addition of both a centrifugation and a washing step may also be appropriate when considering the design of in vitro culture experiments for developmental analysis and stage-specific gene expression as this appears to increase the synchronicity of early developmental stages.
MATERIALS AND METHODS
Source of oocysts
Both C. parvum Swiss cattle C26 and C. parvum Iowa isolates were used in these studies. C. parvum cattle isolate (Swiss cattle C26) oocysts, originally obtained from the Institute of Parasitology, Zurich, were purchased from the Department of Veterinary and Biomedical Sciences, Murdoch University, Perth, Australia. C. parvum cattle isolate (Iowa) oocysts were obtained from Waterborne Inc. New Orleans, LA, USA. Both isolates were used for all centrifugation and excystation experiments. On receipt, infectivity of each oocyst batch was determined using a cell culture-Taqman PCR assay (Keegan et al. Reference Keegan, Fanok, Monis and Saint2003) and, unless where otherwise stated, all experiments using oocysts were carried out within 12 weeks of receipt. Oocyst counts were performed using both flow cytometry (Vesey et al. Reference Vesey, Griffiths, Gauci, Deere, Williams and Veal1997) and fluorescence microscopy (Keegan et al. Reference Keegan, Fanok, Monis and Saint2003). An Olympus BX60 microscope was used for examination of samples stained with EasyStain (BTF, Sydney, New South Wales, Australia) at 200 or 400 times magnification.
Oocyst excystation protocol
C. parvum oocysts were pre-treated to promote excystation using a standard procedure. Oocysts were incubated in 1 ml of acidified water (pH 2·4)-trypsin (0·025% (wt/vol)) for 20 min at 37°C, centrifuged (10 min at 1800 g) and re-suspended in 1 ml of supplemented RPMI 1640 medium (37°C) (Sigma, Australia) (Keegan et al. Reference Keegan, Fanok, Monis and Saint2003) ready for subsequent experimentation. Excystation rates were determined for each oocyst batch using flow cytometry (Vesey et al. Reference Vesey, Griffiths, Gauci, Deere, Williams and Veal1997). In a number of experiments a modified excystation technique was substituted which involved oocysts being incubated in the same solution for 20 min at 30°C, followed by centrifugation (10 min at 1800 g) and re-suspension in 1 ml of supplemented RPMI 1640 medium at room temperature before in vitro infections and flow cytometric studies.
Excystation measurements using flow cytometry
Oocysts pre-treated using both excystation procedures were re-suspended in 1 ml of maintenance medium at 37°C for time-periods ranging from 0 through to 2 h. Oocysts were centrifuged (10 min at 1800 g), supernatant removed down to 100 μl and resuspended in 390 μl of PBS with 10 μl of monoclonal antibody EasyStain (BTF, Sydney, New South Wales, Australia). The monoclonal antibody detects only the oocyst and does not react with the sporozoites. Samples were incubated for 15 min in the dark at RT. Stained oocysts were then analysed by flow cytometry for excystation using a procedure described previously (Vesey et al. Reference Vesey, Griffiths, Gauci, Deere, Williams and Veal1997). Briefly, flow cytometry was performed using a Becton Dickinson (San Jose, USA) FACSCalibur flow cytometer. Logarithmic signals were used for all parameters and the forward light scatter detector was set to E00, the side light scatter detector set to 335 V, and the green fluorescent detector set to 350 V. Each sample was collected for a defined time-period of 2 min with a minimum of 3000 events collected. To ensure that non-oocyst particles were removed from the data analysis, immunofluorescently stained particles were gated on a plot of side scatter versus green fluorescence data. That region was subsequently analysed by a scatter plot of forward versus side scatter data. Those excysted oocysts were identified by their relative decrease in forward light scatter compared to intact oocysts (Vesey et al. Reference Vesey, Griffiths, Gauci, Deere, Williams and Veal1997).
Cell-culture-Taqman infectivity assay
Oocyst concentrations were determined by microscopy and flow cytometry. In total, 10 000 oocysts were used for infection of each cell monolayer. The HCT-8 cell line was seeded into 24-well plates (Greiner Bio-One CELLSTAR® 662 160) at 5×105 cells/well and grown until confluent over a 48 h incubation period before infection. In vitro culturing, cell-culture infection of the HCT-8 line, and DNA extraction from the infected monolayer were as previously described (Keegan et al. Reference Keegan, Fanok, Monis and Saint2003). Real-time PCR and the preparation of DNA standards for the quantification of cell-culture infection were as previously described (King et al. Reference King, Keegan, Monis and Saint2005). Normalized infectivity was calculated using the equation; Infectivity of the sample=(Taqman PCR result of the modified treatment/Taqman PCR result of the standard treatment) × 100.
Cell culture focus detection method
Oocysts were applied to the cell-culture monolayer after pre-treatment using either the standard or modified excystation protocol. Unless stated otherwise, 100 oocysts were applied to each individual well of a LAB-TEK II (Nalge Nunc International) chamber slide. The HCT-8 cell line was seeded at 4×105 cells/well and grown until confluent over a 48 h incubation period before infection. In vitro culturing and cell-culture infection of the HCT-8 line were as for the cell-culture-Taqman infectivity assay (Keegan et al. Reference Keegan, Fanok, Monis and Saint2003). After culture, supplemented medium was removed by aspiration before each well was fixed with 200 μl of methanol for 10 min at RT. Methanol was then removed by aspiration, 200 μl of rehydrating blocking buffer were added (phosphate-buffered saline, pH 7·5 (PBS) with 5% BSA) and incubated for 30 min at RT. Buffer was then removed and 100 μl of 1× Sporo-Glo (Waterborne) antibody applied before incubation for 30 min in the dark on an orbital platform. Antibody was removed and wells washed 4 times with 1×PBS. LAB-TEK well partitions were removed from the slide before the addition of 5 μl of mounting medium per well (BTF, Sydney, New South Wales, Australia). Wells were sealed with glass cover-slips before analysis using a fluorescence microscope.
Foci of infection were counted under blue light at 100× magnification (with verification at 200×where appropriate), using a Nikon Eclipse 80i fluorescence microscope. Images were captured for measurement with a Nikon DS-5M-L1 digital camera and measurements made from the stored images using the NIS-Elements image analysis software (v 3.10). To minimize bias in the selection of foci for measurement, 3–5 foci were photographed from each of the 8 wells, in the order in which they were encountered, avoiding only foci that touched or overlapped the edge of the well or where the cell sheet was disturbed. The number of fluorescent bodies in each focus was counted, without attempting to judge which life-cycle stage they represented. The area infected was measured as the sum of the distinguishable infected ‘zones’ in the focus rather than as a single polygonal figure.
Statistics
Unless otherwise stated the statistical analysis conducted was a Student's t-test and the level of significance was set at a P value of <0·01. The mean and standard deviation were also determined.
RESULTS
To investigate Cryptosporidium excystation by flow cytometric analysis, oocysts were treated using a standard excystation procedure, then subjected to varying time-courses in supplemented RPMI medium before staining with an antibody specific to an oocyst wall protein. Stained oocysts were then subjected to flow cytometric analysis (Fig. 1), which revealed the majority of oocysts excysting by 30 min after the initial excystation treatment. Excysting and excysted oocysts (R2) were identified by their relative decrease in forward light scatter compared to the intact oocysts (R1) (Fig. 1 plots A1-A3) (Vesey et al. Reference Vesey, Griffiths, Gauci, Deere, Williams and Veal1997). The rapid excystation of sporozoites from Cryptosporidium oocysts prompted us to investigate parameters that may enhance the timing of sporozoite contact with a target cell, particularly variation in the volume of medium in which pre-treated oocysts were applied to the monolayer and addition of centrifugation of oocysts onto the monolayer.
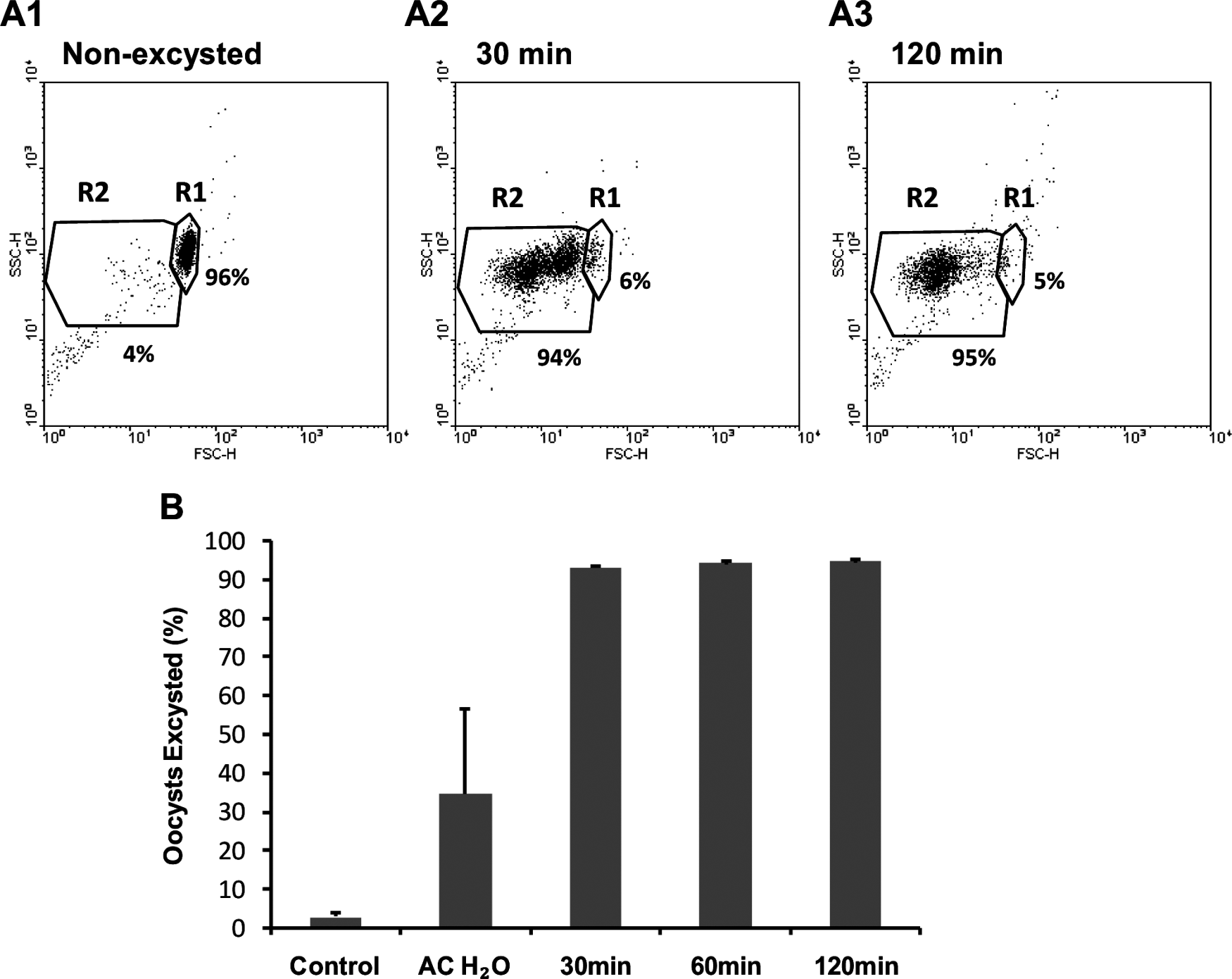
Fig. 1. Time-course experiments of Cryptosporidium parvum excystation using a standard protocol. Representative dot plots of the flow cytometric analysis of unexcysted oocysts (A1) and excysted oocysts (A2 and A3) stained with an antibody specific to an oocyst wall protein which does react with the sporozoites. Region 1 (R1) has been previously identified as the region representing non-excysted oocysts, while region 2 (R2) consists of excysted and excysting oocysts. Intact oocysts not taken through the standard excystation method were used as non-excysted controls. Oocysts taken through the standard excystation method (acidified water treatment at 37°C) were incubated at 37°C in RPMI-supplemented medium for defined time-periods (30 min, 60 min and 120 min) before cytometric analysis (B). Each data point is the mean value from 3 independent experiments and the error bars indicate the standard deviations between experiments.
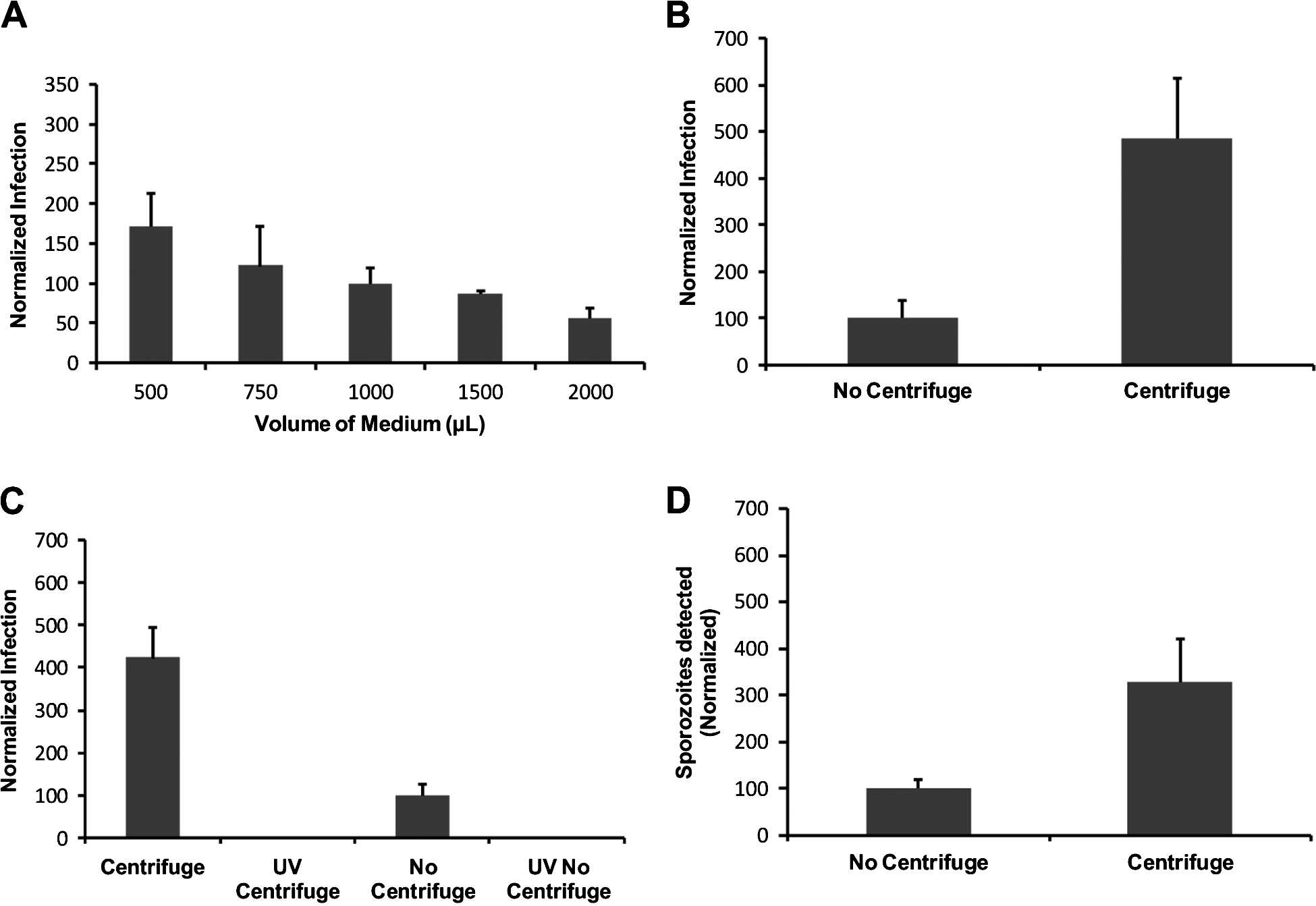
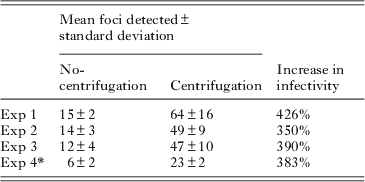
Reducing the initial volume of the medium in which the pre-treated oocysts were applied to cell monolayers resulted in increased in vitro infectivity, as determined by the cell-culture Taqman assay (Single factor ANOVA, P<0·01) (Fig. 2A). However, centrifugation of pre-treated oocysts onto the cell monolayer resulted in superior increases in infectivity in comparison to the no-centrifuge control (approx. 5-fold) (P<0·01) as determined by the cell-culture Taqman assay (Fig. 2B). To confirm that the increases measured by QPCR were indeed reflective of increased infectivity and not increased QPCR signal from embedded oocysts on the HCT-8 monolayer that were not removed during the washing steps, the experiment was repeated with an additional control, consisting of oocysts inactivated by UV-C (40 mJ/cm2 dose) (Fig. 2C). The centrifuged treatment again exhibited a comparable increase in infectivity (approx. 4-fold) in comparison to our standard assay format (P<0·01). However, no significant QPCR signal was evident for the UV treatments, suggesting that increases in QPCR resulted from genuine increases in cell-culture infectivity. Analysis of infected monolayers at 2 h post-infection confirmed that this increase in infectivity resulted from an increase in initial sporozoite attachment that was comparable to increases witnessed in infectivity (approx. 3 to 4-fold) (P<0·01) (Fig. 2D). To further confirm the positive affect of centrifugation on parasite in vitro culture we also utilized the focus detection method. Four individual experiments were performed with centrifugation treatments, each demonstrating an approximately 4-fold increase in infectivity above the standard no centrifugation controls regardless of oocyst age (P<0·01) (Table 1).
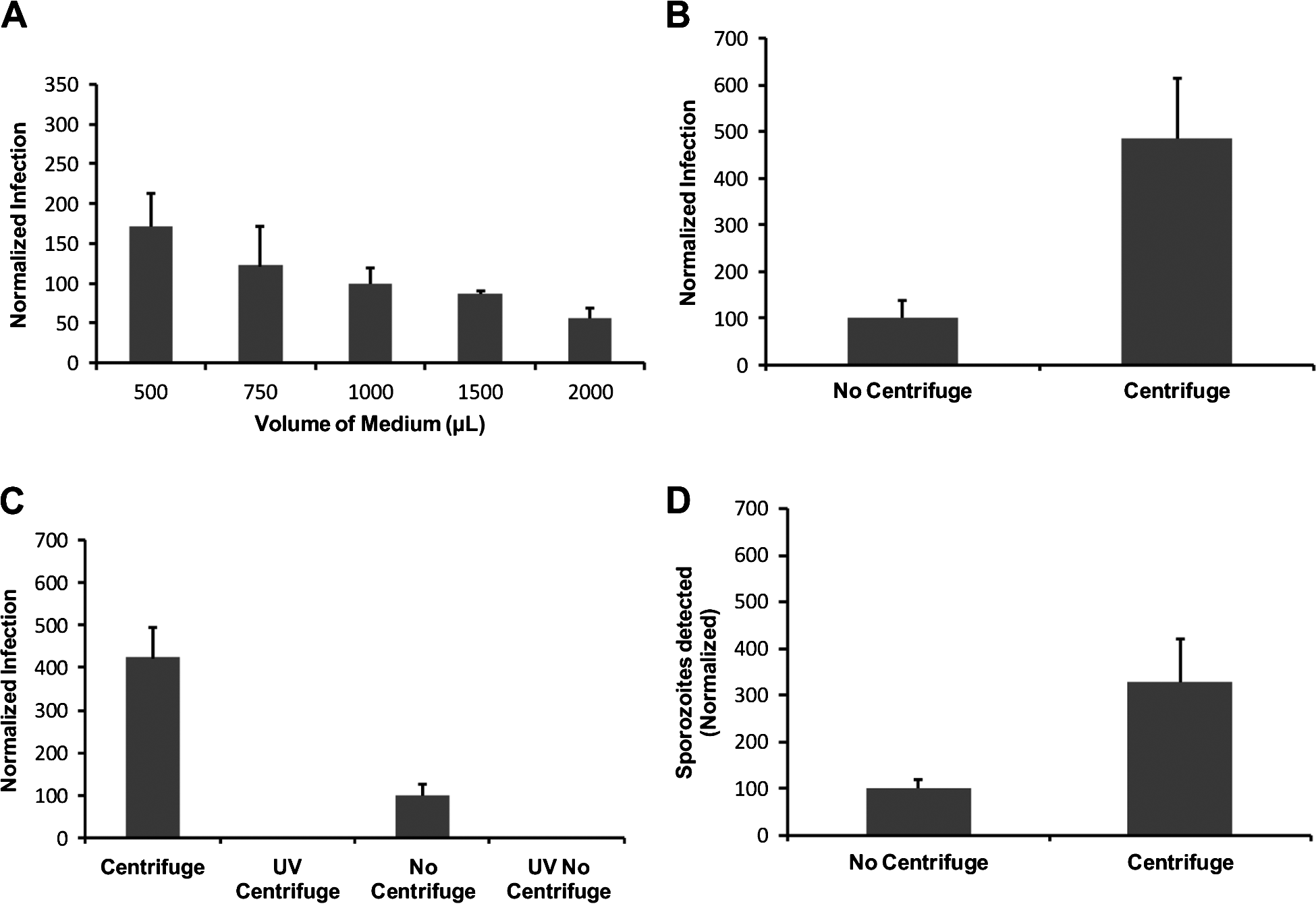
Fig. 2. Representative experiments of variations in the volume of medium containing oocysts applied to the HCT-8 monolayer and the incorporation of centrifugation for Cryptosporidium parvum in vitro infection. Oocysts were pre-treated using standard excystation protocols before application to HCT-8 cell monolayers. For variation in the addition of medium (A), oocysts were applied to the monolayer in varying amounts of supplemented RPMI medium. After incubation for 2 h at 37°C, the volume of medium in individual infected monolayer wells was made up to a total volume of 2 ml and plates were incubated for a further 2 days. Infectivity was expressed as a percentage of the standard assay format (1 ml). For the centrifugation treatments (B) and (C), pre-treated oocysts were spun onto the monolayer at 408 g for 5 min. Control treatments without centrifugation were left on the bench for the duration of the spins. For the UV-C (254 nm) treatments (C), a 40 mJ/cm2 dose was applied to the oocysts using a collimated beam. Infectivity was calculated using a cell-culture-Taqman PCR assay and expressed as a percentage of the no-centrifuge treatments (B–D). With the exception of the sporozoite attachment assay, infected monolayers were incubated for 48 h at 37°C. Sporozoite attachment/invasion was calculated using a cell-culture-Taqman PCR assay and expressed as a percentage of the no-centrifuge control after a 2-h incubation period at 37°C. Error bars indicate standard deviations (n=6).
Table 1. Infectious foci detected for centrifuge and no-centrifuge treatments

* Experiment 4 used oocysts greater than 6 months old, while for Exps 1–3 oocysts were less than 3 months old. For each experiment a minimum of 8 cell-culture replicates were undertaken for each treatment.
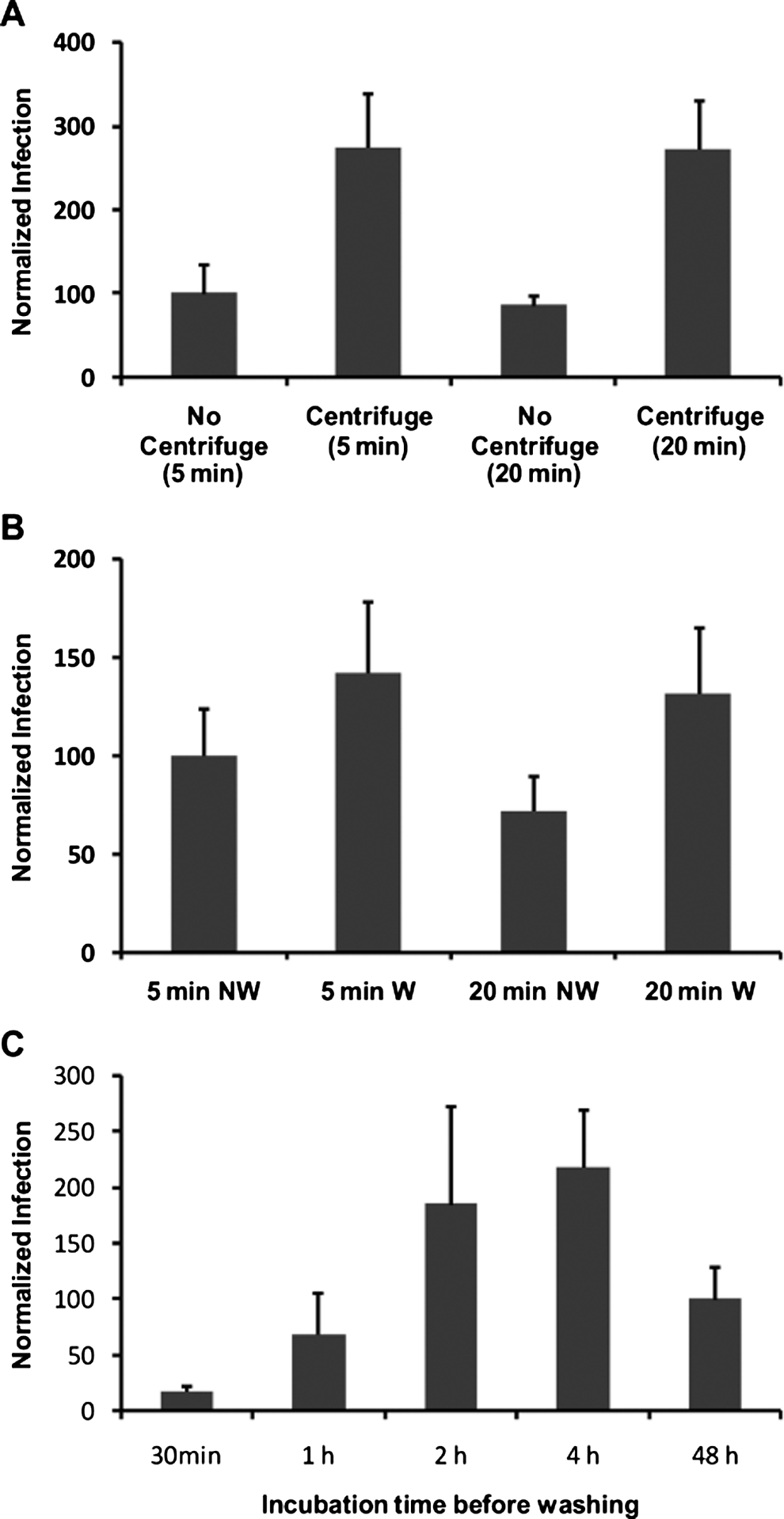
The length of the centrifugation period was increased to examine whether all potentially infectious oocysts were in close enough contact to infect the monolayer (Fig. 3A). However, increasing the length of the centrifugation period did not appear to further increase infectivity, suggesting that a short centrifugation period was sufficient to bring oocysts and sporozoites in close enough proximity to the cell monolayer to maximize infection. A washing step was incorporated in an attempt to increase the synchronicity of early infection by removing those sporozoites unable to successfully attach/invade the cell monolayer. The incorporation of washing the monolayer at 2 h post-infection resulted in a further positive increase in infection above that for centrifugation alone (P<0·01) (Fig. 3B). Additionally, the time at which the monolayer was washed after initial infection was investigated as this step could result in sporozoites successfully attached but not yet encapsulated by the host cell membrane being accidently removed during washing steps, negatively impacting on in vitro infectivity. Therefore an experiment to examine the effect of washing the monolayer at various time-points post-infection was conducted. The optimal time for washing the monolayer was determined to be between 2 and 4 h post-infection (Fig. 3C). Washing the monolayer earlier reduced infection, while not washing the monolayer until 48 h post-infection (standard assay format) also resulted in inferior infection in comparison to the 2 and 4 h time-points (P<0·01).

Fig. 3. Representative experiments of the effect of washing and centrifugation on Cryptosporidium parvum in vitro infectivity. Oocysts were pre-treated using standard excystation protocols before application in supplemented RPMI medium to a HCT-8 cell monolayer. For the centrifugation treatments, pre-treated oocysts were spun onto the monolayer at 408 g for either 5 or 20 min (A). Paired no-centrifuge treatments were left on the bench for the duration of the spin. Pre-treated oocysts were also centrifuged onto the monolayer at 408 g for either 5 or 20 min and infected monolayers were either washed (W) with PBS at 2 h post-infection and the medium replaced, or they were not washed (NW) (B). Pre-treated oocysts were also centrifuged onto the monolayer at 408 g for 5 min and infected monolayers were subjected to various incubation periods at 37°C before washing with PBS and replacement of medium. Infected monolayers were subsequently incubated until a total incubation period of 48 h (A–C). Infectivity was calculated using a cell-culture-Taqman PCR assay and expressed as a percentage of either the no-centrifuge control (5 min) (A), 5 min NW control (B) or the 48 h incubation period no-wash control (C). Error bars indicate standard deviations (n=6).
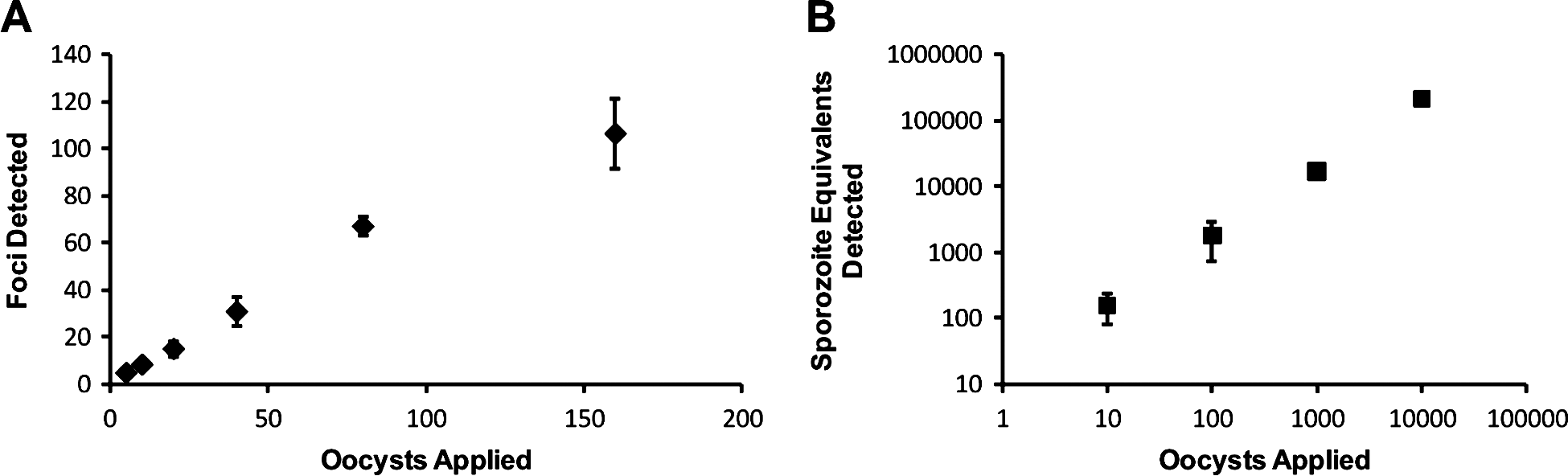
While centrifugation of oocysts onto the monolayer resulted in positive increases for in vitro culture infectivity, it was necessary to establish that the cell monolayer did not become saturated with life-cycle stages by effectively increasing the number of infectious oocysts in contact with the monolayer. Therefore, standard curves to examine the relationship between applied oocysts and infectivity were constructed for both the focus detection method (Fig. 4A) and the cell culture Taqman assay (Fig. 4B). A linear relationship was observed between foci detected and oocyst number over a range of 5–160 oocysts (R2=0·98). While for the Taqman assay the range was more dynamic with a strong linear relationship demonstrated (R2=0·99) between 10 and 10 000 oocysts when applied to an individual monolayer.
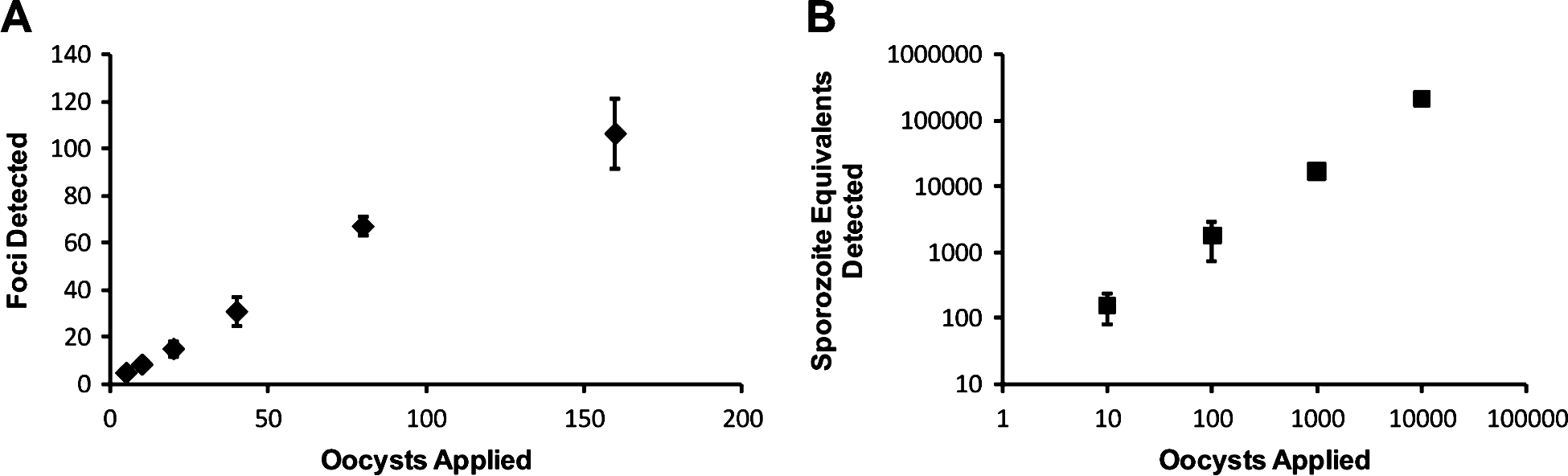
Fig. 4. Establishment of the relationship between oocyst numbers applied to cell-culture monolayers and the number of foci and sporozoite equivalents enumerated. Cryptosporidium parvum oocysts were pre-treated using standard excystation protocols before centrifugation in supplemented RPMI medium onto a HCT-8 cell monolayer. Varying numbers of oocysts were applied to cell monolayers for the focus detection method (A) and the cell-culture-Taqman PCR assay (B). Error bars indicate standard deviations (focus detection n=8) (Taqman PCR assay n=6).
Centrifugation and washing of the cell monolayer was therefore incorporated into our standard assay format in our attempt to identify in vitro stages of parasite amplification using QPCR. A series of 5 independent experiments was undertaken in which the cell monolayer was infected with oocysts, the monolayer washed at 2 h post-infection and then infected monolayers recovered at different time-points during a 48 h incubation period. No amplification in parasite numbers was detectable until 12–14 h post-infection (Fig. 5). The increase in amplification was a 2 to 8-fold increase above the initial numbers of sporozoites quantified as attaching/invading the cell monolayer at 2 h. This suggested the formation of merozoites in our in vitro cell-culture system within this time-frame, in agreement with previously reported work (Hijjawi et al. Reference Hijjawi, Meloni, Morgan and Thompson2001). However by 16 h post-infection, substantial increases in parasite amplification in cell culture were apparent (Fig. 5), with further large increases accompanied by longer incubation periods, suggesting that the generation time between merozoites and subsequent parasite life cycles is greatly reduced compared with the initial time required between attachment/invasion of sporozoites and generation of the first merozoites.
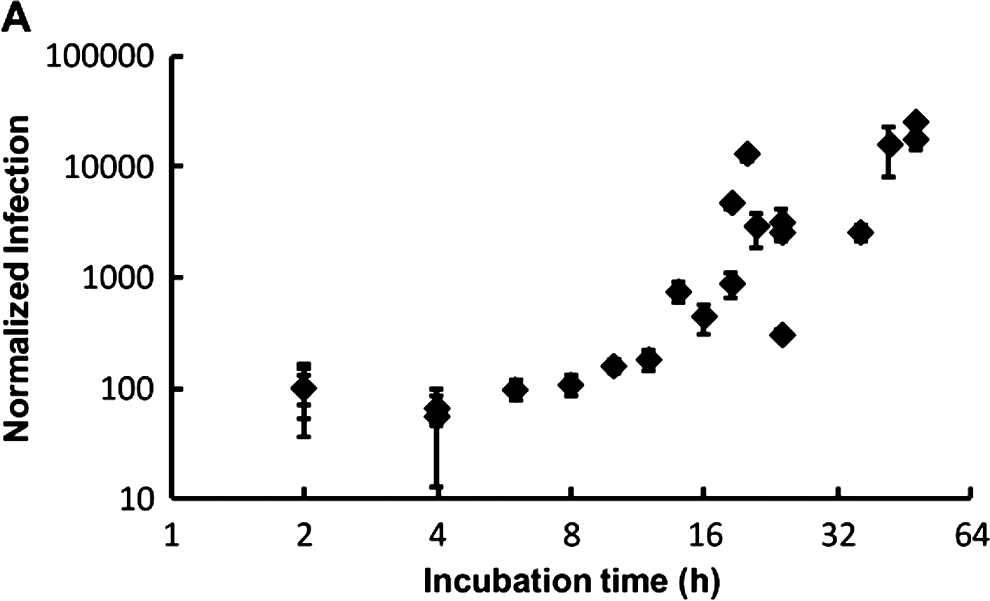
Fig. 5. Time-course of Cryptosporidium parvum amplification in cell culture over 48 h. Oocysts were pre-treated using standard excystation protocols before centrifugation in supplemented RPMI medium onto a HCT-8 cell monolayer. Infected monolayers were washed with PBS at 2 h, the media replaced, then incubated at 37°C for defined time-periods before the infected monolayer was harvested. Infectivity was calculated using a cell-culture-Taqman PCR assay and expressed as a percentage, relative to the sporozoites that had attached/invaded the cell monolayer at 2 h post-infection. The figure represents data generated from 5 separate experiments. A 2 h time-point was included for each experiment. Each data point represents the mean infectivity from an individual experiment and error bars indicate standard deviations for cell culture infectivity of that experiment (n=6).
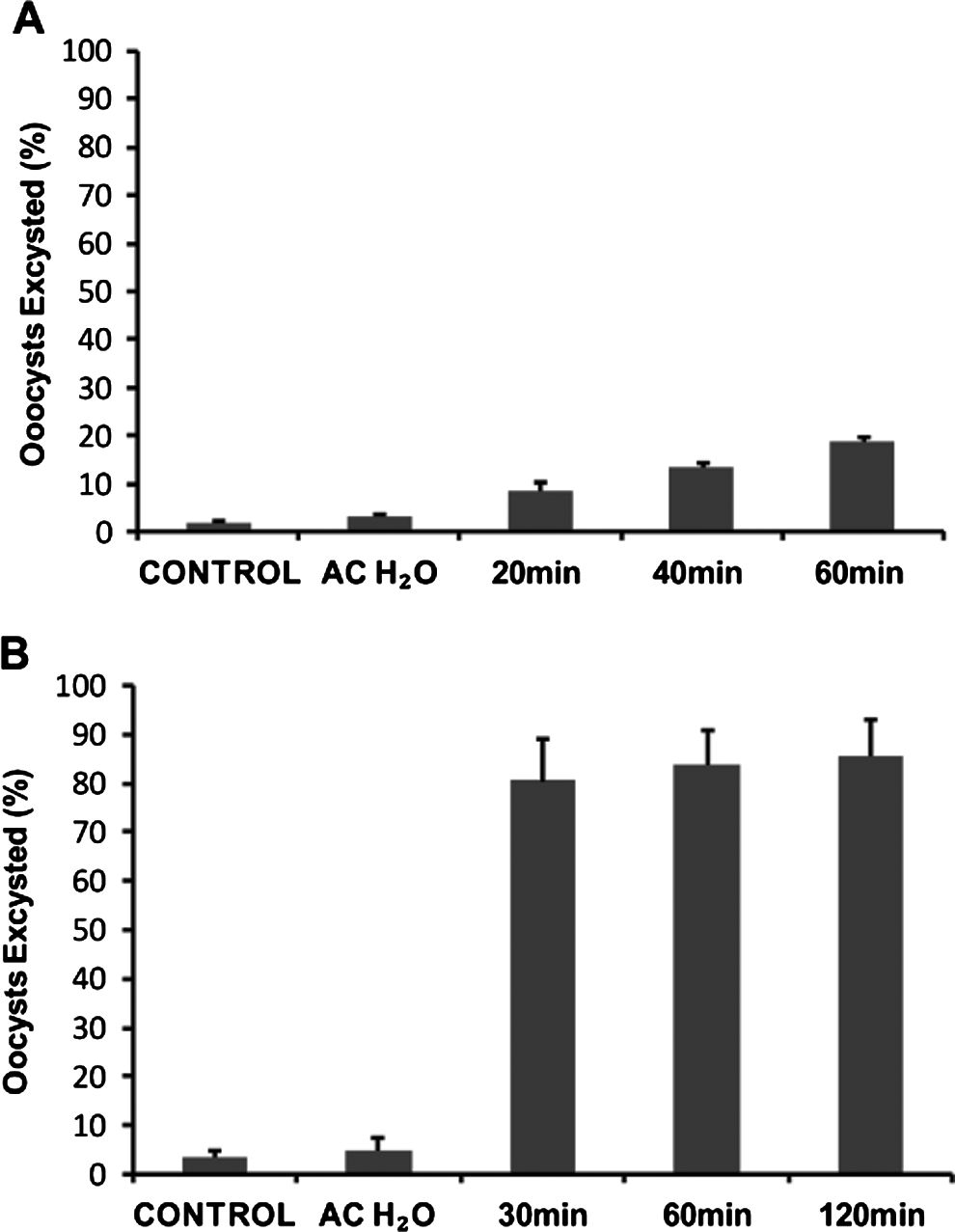
We used the focus detection method at various time-points to compare with data generated from the QPCR analysis of in vitro parasite amplification. A focus is theoretically defined as an area of infection formed by a single infectious oocyst (Rochelle et al. Reference Rochelle, Ferguson, Johnson and De Leon2001). However, it was obvious from our standard excystation methodology that a large percentage of oocysts could excyst before they contact the cell monolayer. This suggested the possibility that multiple foci may develop from the sporozoites from a single oocyst, potentially confusing the enumeration of foci. We therefore examined the possibility of slowing the initial rate of oocyst excystation to limit the likelihood of sporozoite release before oocysts are in contact with the monolayer. A number of different variables were trialed for their effect on the rate of oocyst excystation. Reducing the temperature of the acidified water pre-treatment of oocysts from 37°C to 30°C in concert with the addition of RT-supplemented medium instead of warm medium greatly reduced the rate of excystation (Fig. 6A). However, once incubated at 37°C in supplemented medium, a rapid rate of excystation was re-established and by the end of a 2 h period the majority of oocysts had excysted (>85%) (Fig. 6B).
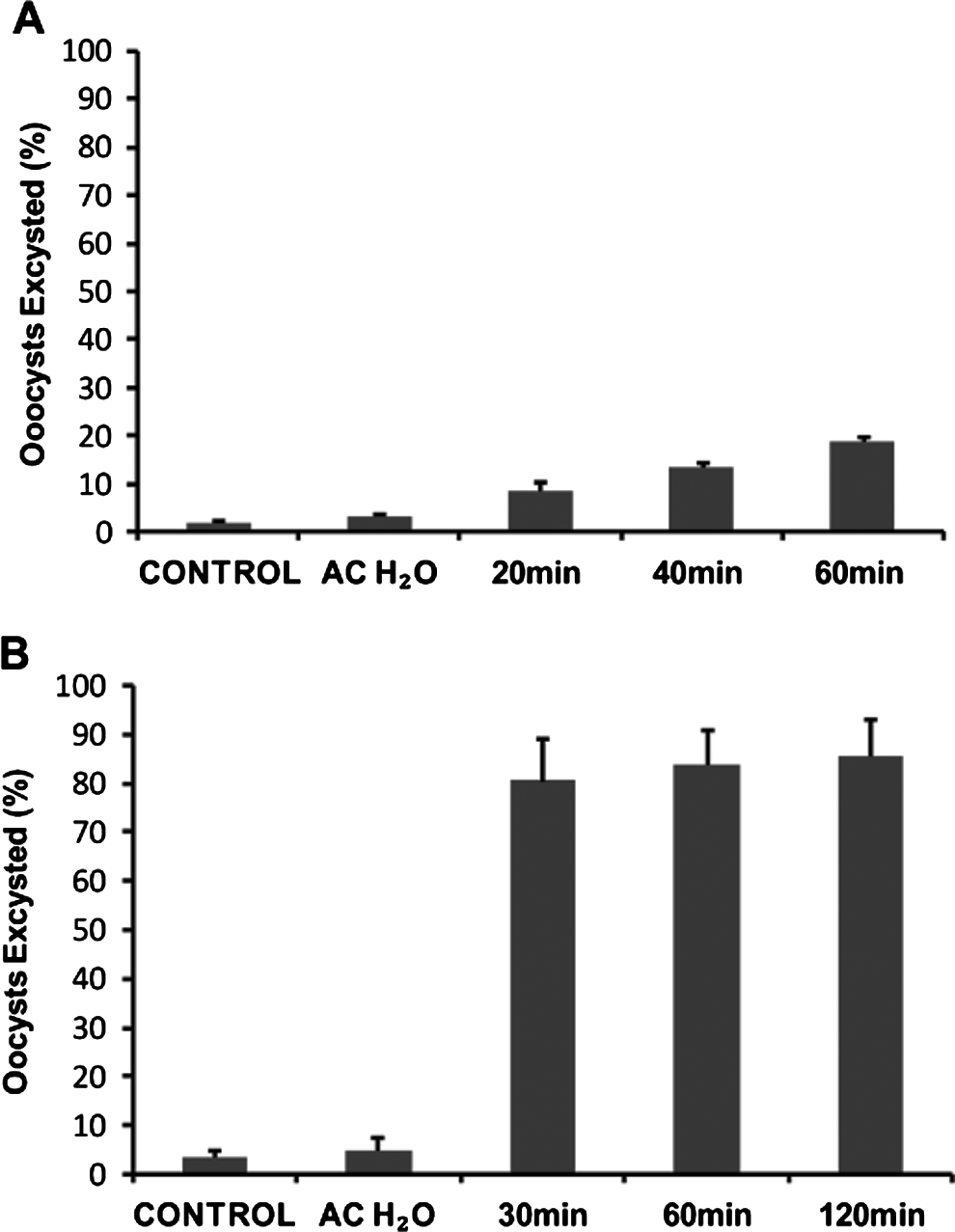
Fig. 6. Time-course experiments of Cryptosporidium parvum excystation, using a modified excystation protocol with alternative subsequent incubation temperatures. Intact oocysts not taken through the excystation method were used as controls. Oocysts taken through the modified excystation method (acidified water treatment at 30°C) were incubated at room temperature in RPMI supplemented medium for defined time-periods (20 min, 40 min and 60 min) before flow cytometric analysis (A). Oocysts taken through the modified excystation method were also incubated at 37°C in RPMI-supplemented medium for defined time-periods (30 min, 60 min and 120 min) before flow cytometric analysis (B). Each data point is the mean value from 3 independent experiments and the error bars indicate the standard deviations between experiments.
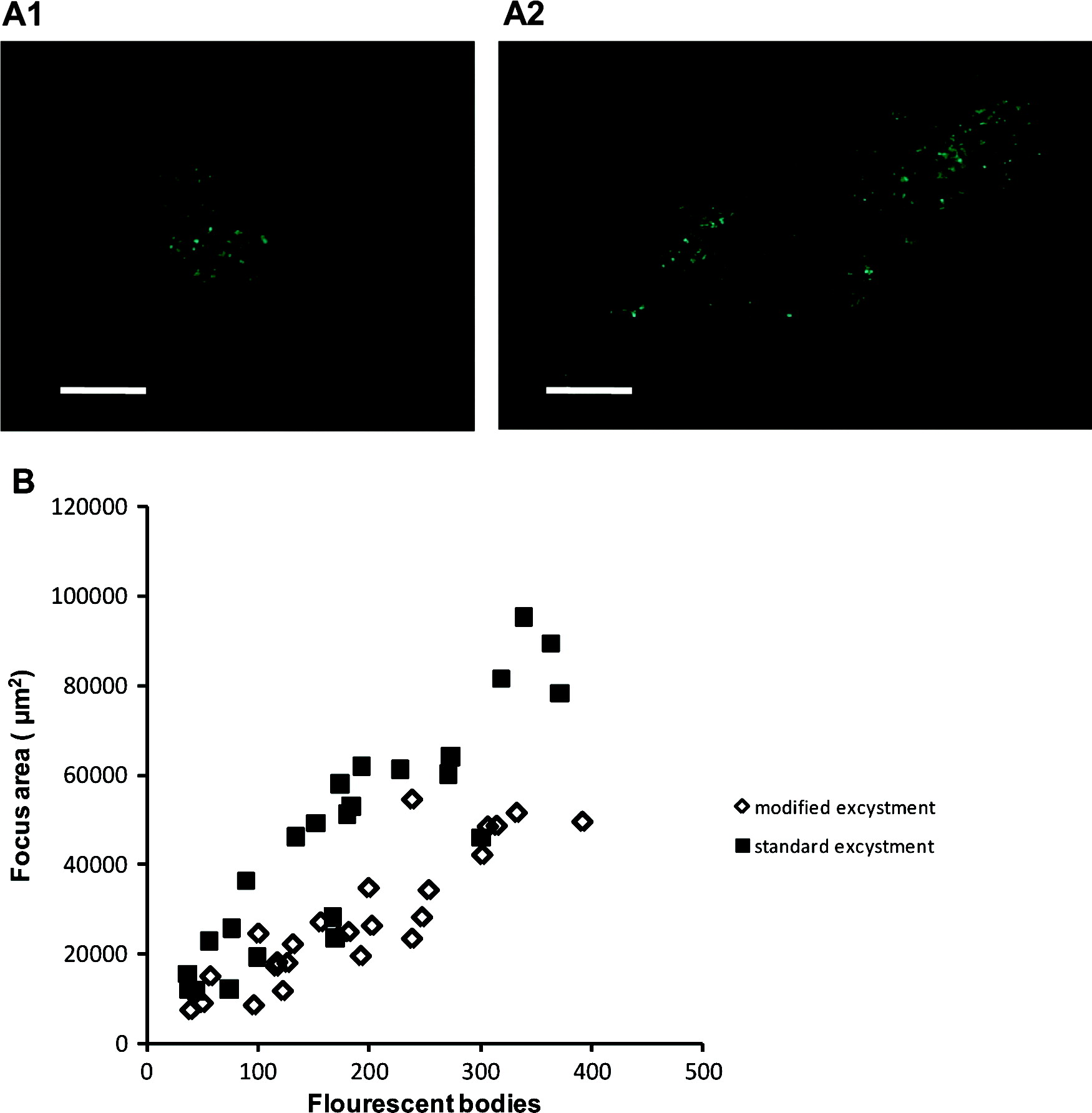
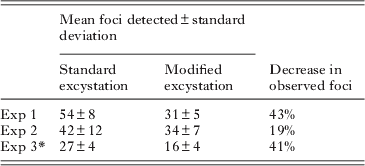
The modified excystation method resulted in significantly fewer foci (P<0·01) for both Exp. 1 and Exp. 3, and fewer (though not statistically significant) in Exp. 2 (Table 2). Foci resulting from both excystment protocols consisted typically of discrete ‘zones’ of infection (commonly 2, occasionally 3 or 4), which were interpreted as being initiated separately by sporozoites released from a single oocyst. Foci were variable in size, consisting of fewer than 40 to more than 350 fluorescent bodies at 48 h (standard protocol – mean 182±105, median 174; modified protocol mean 188±95, median 182; n=25). The area of HCT-8 cells infected correlated positively with the number of fluorescent bodies, and was greater for the standard protocol (median 4·9×104 μm2, r=0·9059) than for the modified protocol (median 2·5×104 μm2, r=0·8909) (Fig. 7). Foci of infection resulting from the modified protocol were therefore considerably more compact (density of life cycle stages per μm2) than those resulting from the standard protocol (P<0·01).
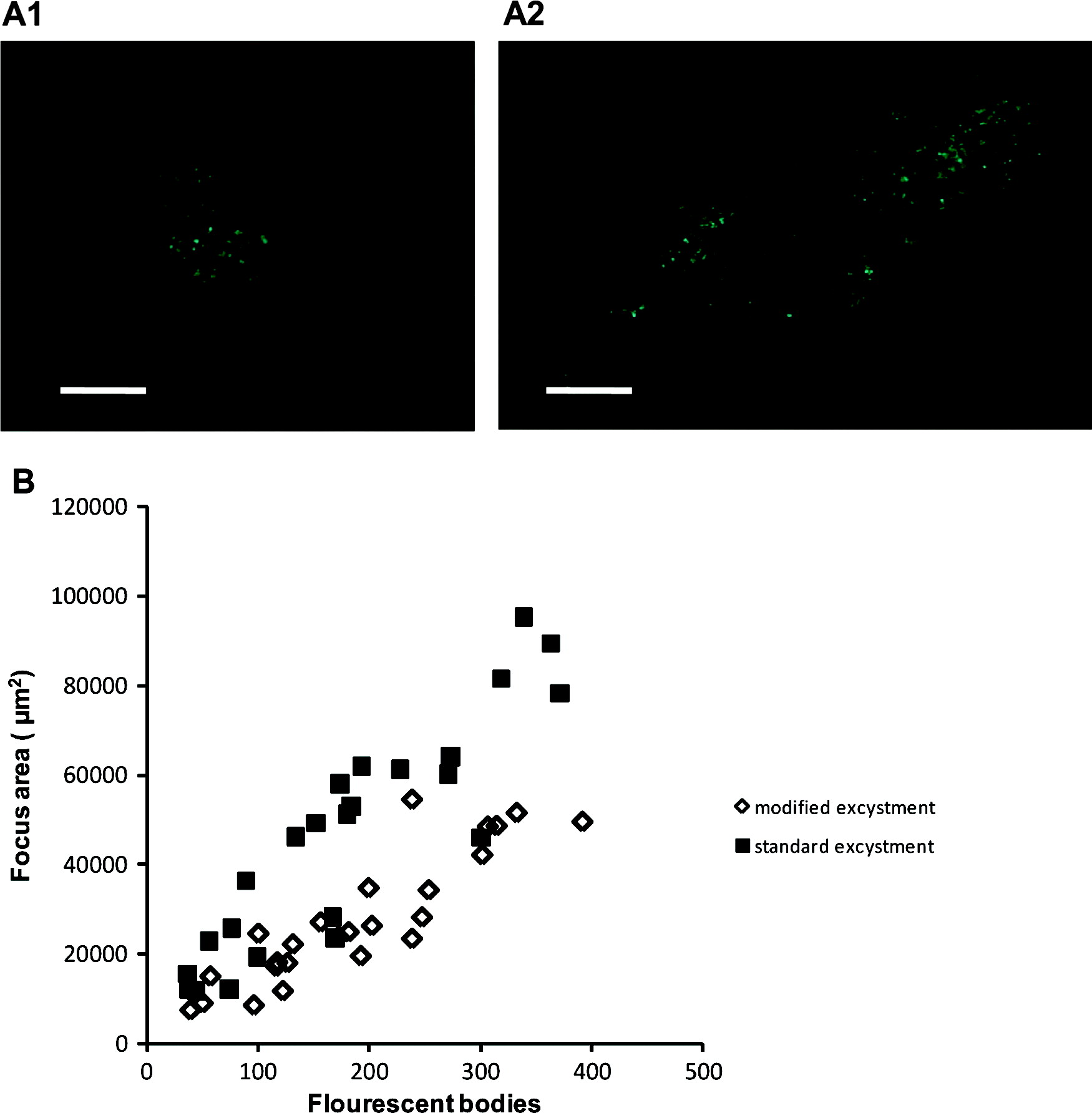
Fig. 7. Representative focus images for the modified excystation methodology (A1) and the standard excystation methodology (A2). Scale bars=100 μm. The numbers of fluorescent bodies within individual foci were counted and the area of infection measured for both treatments in order to express a relationship between focus area and life-cycle stages (B).
Table 2. Infectious foci detected using the standard and modified excystation treatments
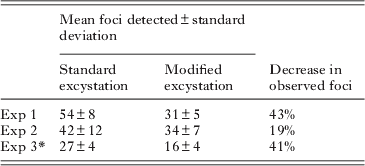
* Experiment 3 used oocysts that were 2 months old, while for Exps 1 and 2 oocysts were less than 1 month old. For each experiment a minimum of 8 cell-culture replicates were undertaken for each treatment.
The modified excystation methodology was therefore utilized in combination with centrifugation of oocysts onto the monolayer and subsequent analysis of different time-periods during a 48-h incubation period using the focus detection method. The focus detection method confirmed a dramatic increase in life-cycle stages after 16 h post-infection as indicated by the QPCR data (data not shown).
DISCUSSION
Together with a number of laboratories worldwide, we employ Cryptosporidium in vitro culture for the detection of infectious oocysts isolated from the environment, to quantify oocyst inactivation by a range of stresses and to study the parasite life cycle at both molecular and morphological levels. However, recent work emphasizing the short-lived nature of sporozoites after excystation and the rapid physiological and biochemical events that occur soon after excystation (King et al. Reference King, Hoefel, Lim, Robinson and Monis2009; Matsubayashi et al. Reference Matsubayashi, Ando, Kimata, Nakagawa, Furuya, Tani and Sasai2010), prompted us to revisit the current design of in vitro parasite culture.
Oocysts have a low settling velocity, resulting from their small size and low specific gravity. This rate has been reported in the literature to range between 0·22 μm/sec and 1 μm/sec (Kim and Corapcioglu, Reference Kim and Corapcioglu2002; Searcy et al. Reference Searcy, Packman, Atwill and Harter2005). Our standard 24-well plate Taqman assay uses a total oocyst suspension volume of 1 ml that results in a medium depth of 5 mm. For the focus detection method the medium depth within the wells is 4 mm. Based on the above settling rates, oocysts near the top of the suspension could take between 1 and 5 h to reach the monolayer. Our flow cytometric studies indicated that, using our standard excystation assay, the majority of sporozoites excyst within 30 min post-excystation treatment. Once excysted, sporozoites go through rapid physiological and biochemical changes that can dramatically reduce their infectivity (King et al. Reference King, Hoefel, Lim, Robinson and Monis2009; Matsubayashi et al. Reference Matsubayashi, Ando, Kimata, Nakagawa, Furuya, Tani and Sasai2010). We therefore postulated that in the current assay format, a majority of the excysted sporozoites in the supplemented medium may not be close enough to the cell monolayer to infect. While excysted sporozoites can actively move by helical gliding at a maximum speed of 5·1 μm/sec (Wetzel et al. Reference Wetzel, Schmidt, Kuhlenschmidt, Dubey and Sibley2005), this movement is substrate dependent and any excysted sporozoites in the medium and not in intimate contact with the monolayer would have limited opportunity to find and infect a compatible cell before extensive apical organelle discharge ensues (Chen et al. Reference Chen, O'Hara, Huang, Nelson, Lin, Zhu, Ward and Larusso2004; King et al. Reference King, Hoefel, Lim, Robinson and Monis2009).
In order to maximize infection, we examined the effect of changing the volume of the medium in which oocysts were applied to the monolayer, as well as the addition of a centrifugation step. Reducing the volume of the medium in which the pre-treated oocysts were applied, as well as centrifugation, made substantial increases to in vitro infectivity, with centrifugation consistently resulting in increases between 3 and 5-fold above the standard treatment for both the focus detection and cell culture Taqman assays. The energy-intensive nature of helical gliding, the finite energy reserves available to sporozoites and their ensuing apical organelle discharge, reinforce the importance of sporozoites excysting in close proximity to a compatible host cell. The incorporation of the centrifugation of oocysts onto the monolayer in most assay formats will therefore help to maximize in vitro infectivity and assay sensitivity.
A previous report by Weir et al. (Reference Weir, Pokorny, Carreno, Trevors and Lee2001) identified that centrifugation of oocysts onto the monolayer increased the rate of infection. While those authors identified more rapid development of foci, they reported no increase in the number of infective foci by 48 h, in contrast to the work presented here. However the authors did demonstrate significant and large differences in foci number between non-centrifuged and centrifuged treatments for the earlier 18 and 24 h incubation periods. Notably the authors reported that they replaced the medium at 24 h for the 48 h incubation period, possibly and inadvertently resulting in a loss of invasive life-cycle stages. Secondly, the authors used a different excystation protocol where oocysts were exposed to sodium hypochlorite before incubation within pre-warmed medium and centrifugation onto the monolayer, possibly delaying the excystation of oocysts and allowing those oocysts in the non-centrifuged treatments more time to settle onto the monolayer before excystation and subsequent infection.
Previous studies employing the focus detection method to determine in vitro infectivity and defining foci by the of criteria of Rochelle et al. (Reference Rochelle, Ferguson, Johnson and De Leon2001) have reported low infectivity of 4–22% even for freshly excreted oocysts (Rochelle et al. Reference Rochelle, Ferguson, Johnson and De Leon2001; Connelly et al. Reference Connelly, Wolyniak, Williamson and Jellison2007). These results are within the range we report when using our standard assay format, which does not include centrifugation of oocysts onto the monolayer. The work presented here demonstrates that oocyst excystation rate in concert with the format of the in vitro assay utilized, notably parameters such as medium volume/depth in which oocyst inoculum is applied, can dramatically affect assay sensitivity. The use of centrifugation of oocysts onto the monolayer may therefore increase assay sensitivity for various assay formats.
In order to use Cryptosporidium-infected cells as material for developmental analysis and stage-specific gene expression, the development of parasite stages needs to be well defined in the in vitro system employed. A particular goal of this work was to define precisely the attachment/invasion of sporozoites and the generation of merozoites in our cell-culture infectivity system so as to enable such future work. However, it has been previously reported in the literature that the Cryptosporidium life cycle in vitro is not synchronous, which could confound future genetic experiments envisaged. In the standard assay format, the majority of excysted sporozoites did not infect the monolayer, reducing the density of infected monolayer material available for any subsequent genetic experiments. These results also raised the possibility that those sporozoites which did not infect may have delayed infection, reducing the synchronicity of infection. Finally, it was also recognized that some sporozoites may be able to attach/invade, but be unable to proceed to later stages of the life cycle, further confusing any genetic investigations.
As discussed above, Weir et al. (Reference Weir, Pokorny, Carreno, Trevors and Lee2001) had already suggested the centrifugation of oocysts onto the monolayer to help alleviate this problem. Combining the rapid excystation of oocysts with centrifugation of oocysts/excysted sporozoites onto the monolayer should therefore not only increase the density of the infected monolayer but synchronize the timing of infection. However, those sporozoites able to attach but not invade may remain, potentially confusing the analysis. In an effort further to increase the synchronicity of early infection, we examined the use of washing steps to remove those sporozoites unable to successfully attach/invade the cell monolayer. Sporozoites can quickly attach to and invade compatible cells; the sporozoite attaches to the membrane of a host epithelial cell by its apical end and is subsequently encapsulated by a parasitophorous vacuole (PV) membrane (Chen et al. Reference Chen, O'Hara, Huang, Nelson, Lin, Zhu, Ward and Larusso2004; Borowski et al. Reference Borowski, Clode and Thompson2008). Previous work has reported that maximal attachment/invasion of sporozoites to a cell monolayer occurs by 2 h post-infection (Chen et al. Reference Chen, O'Hara, Huang, Nelson, Lin, Zhu, Ward and Larusso2004; Feng et al. Reference Feng, Nie, Sheoran, Zhang and Tzipori2006), and this was chosen as the initial time-point in our investigation. Subsequent washing experiments confirmed that maximal attachment occurs between 2 h and 4 h post-infection of the monolayer. Washing the monolayer earlier resulted in inferior infection suggesting that sporozoites, which would otherwise go on to successfully infect the monolayer, were being washed away at these time-points.
Unexpectedly, when the cell monolayer was washed at 2 and 4 h post-infection, in vitro infectivity increased compared to the standard assay format. We can think of a number of possible explanations for this increased infectivity. Firstly, sporozoites that do not successfully infect the monolayer may lyse and release inhibitory metabolic products that affect the growth of other successfully infecting zoites and subsequent life-cycle stages. Removing these sporozoites by washing could possibly have a beneficial effect on in vitro infectivity. Secondly, sporozoites able to attach to a host cell but not successfully invade may prevent subsequent life-cycle stages from infecting that cell by blocking the binding sites for infection. Any such sporozoites attached to the monolayer would greatly reduce infection by other life-cycle stages. However, washing may remove these sporozoites before the emergence of other life-cycle stages, freeing such attachment sites. Finally, washing the monolayer after maximal sporozoite attachment/invasion could remove other organisms such as bacteria and fungi, present in the oocyst suspension but not killed by antibiotics in the medium. Such organisms may inhibit parasite replication by the production of inhibitory compounds and/or use of available resources in the supplemented medium needed for healthy maintenance of the cell monolayer.
Our results suggest that washing the monolayer as early as 2 h post-infection had no detrimental effect on infectivity and was beneficial to in vitro parasite growth. This is in agreement with other reports in the literature demonstrating that maximal sporozoite attachment/invasion occurs around 2 h post-inoculation of the monolayer (Chen et al. Reference Chen, O'Hara, Huang, Nelson, Lin, Zhu, Ward and Larusso2004; Feng et al. Reference Feng, Nie, Sheoran, Zhang and Tzipori2006). A number of authors have reported washing the monolayer after initial inoculation of sporozoites; however, it does not appear to have been universally adopted. In a number of experiments subsequently conducted using different oocyst batches (data not presented) we have found that increases in infectivity due to washing, while always positive, can vary considerably, signifying that the reasons behind increases in infectivity may vary as well. This suggests that the presence of other contaminating organisms/chemicals in the oocyst suspension may be a key factor; however, it is probable that increased infectivity is a combination of the factors above, and others we have yet to consider. While washing the cell monolayer may be a prudent practice for experiments aimed at trying to synchronize early life-cycle stage development, we are yet to be convinced that it is appropriate for all applications, especially the detection and enumeration of infectious oocysts from the environment, where slowly excysting oocysts may be washed away at 2 h post-inoculation.
The use of centrifugation and washing of the infected monolayer enabled us to use quantitative real-time PCR (QPCR) to define when Cryptosporidium sporozoites attach/invade the cell monolayer and to identify the timing of initial generation of merozoites in our in vitro system. The time-course of amplification of ‘parasite life cycles’ in our in vitro system should prove helpful in pin-pointing particular times for the production of material for future experiments encompassing differential display, expression analysis and investigation of life-cycle stages.
As previously discussed, oocyst excystation rate can influence assay sensitivity and may also influence data interpretation, depending on the assay format employed. For the focus detection method, an individual focus of infection is theoretically defined as the area infected by sporozoites that excyst from an individual oocyst, which then form a cluster of intracellular life-cycle stages (Rochelle et al. Reference Rochelle, Ferguson, Johnson and De Leon2001). Our standard excystation technique, however, results in a considerable percentage of oocysts releasing their sporozoites before coming into contact with the cell monolayer. By tailoring the excystation rate through varying the initial acid temperature treatment, we were able to more precisely control excystation rate thereby increasing the probability that a single focus of infection would be a reflection of the sporozoites from an individual oocyst. Importantly, we demonstrated that the use of a particular excystation methodology can significantly overestimate the number of infective oocysts present in a sample. Therefore, if the focus detection method is to be utilized, the probability of sporozoites from an individual oocyst infecting a more circumscribed area of the cell monolayer can be maximized by the use of centrifugation in combination with an excystation technique that will quickly release oocysts once they are in contact with the monolayer.
The work presented here reinforces the importance of understanding assay design in order to maximize both sensitivity and synchronicity of in vitro Cryptosporidium infection. It raises significant implications for the centrifugation of oocysts onto cell monolayers, especially in concert with the excystation methodology employed, because these can dramatically affect assay sensitivity as well as interpretation. This may be of acute importance for detection of low numbers of infectious oocysts in drinking water systems and re-use waters when using techniques such as the focus detection method.
ACKNOWLEDGEMENTS
We are grateful to Renae Phillips, Stella Fanok, Shailaja Pannersilvam and Elsa Soubiran for excellent technical assistance. We thank the South Australian Water Corporation, the Water Research Foundation (Water RF) and the Victorian Smart Water Fund for financial, technical and administrative assistance in funding this work. Mention of trade names or commercial products does not constitute Water RF endorsement or recommendations for use. Similarly, omission of products or trade names indicates nothing concerning Water RF's position regarding product effectiveness or applicability. The comments and views detailed herein may not necessarily reflect the views of the Water RF, its officers, directors, affiliates or agents.