INTRODUCTION
Cryptic species, or genetically distinct lineages that have previously been classified as a single nominal species due to their superficially indistinguishable morphological characteristics, are becoming increasingly recognized as an important issue for the study of ecology and evolutionary biology (Bickford et al. Reference Bickford, Lohman, Sodhi, Ng, Meier, Winker, Ingram and Das2006). Beyond the obvious implications it holds for obtaining an accurate inventory of existing biodiversity, the failure to recognize cryptic species in medically, economically or ecologically important organisms can have serious negative or costly consequences for the development of biological control measures (Bidochka et al. Reference Bidochka, Kamp, Lavender, Dekoning and de Croos2001; Walter and Campbell, Reference Walter and Campbell2003; Rafter et al. Reference Rafter, Gillions and Walter2008), the monitoring and control of human parasites and potential zoonoses (Cepicka et al. Reference Cepicka, Kutišova, Tachezy, Kulda and Flegr2005; Pringle et al. Reference Pringle, Baker, Platt, Ware, Latgé and Taylor2005; Saijuntha et al. Reference Saijuntha, Sithithaworn, Wongkham, Laha, Pipitgool, Tesana, Chilton, Petney and Andrews2007), the management of agricultural and aquaculture pathogens (Beauchamp et al. Reference Beauchamp, Gay, Kelley, El-Matbouli, Kathman, Nehring and Hedrick2002; Skobgaard et al. Reference Skobgaard, Bødker and Rosendahl2002), and detecting the presence of invasive species (Geller et al. Reference Geller, Walton, Grosholz and Ruiz1997; Geller, Reference Geller1999; Miura et al. Reference Miura, Torchin, Kuris, Hechinger and Chiba2006).
In the last few years certain taxa of trematodes that were previously identified as a single species based on morphological features have been shown to consist of many genetically distinct cryptic species (e.g., Donald et al. Reference Donald, Kennedy, Poulin and Spencer2004; Miura et al. Reference Miura, Kuris, Torchin, Hechinger, Dunham and Chiba2005; Saijuntha et al. Reference Saijuntha, Sithithaworn, Wongkham, Laha, Pipitgool, Tesana, Chilton, Petney and Andrews2007). This coincides with an increase in the use of genetic markers to identify cryptic species complexes and resolve taxonomic questions among parasitic platyhelminths (Vilas et al. Reference Vilas, Criscione and Blouin2005; Nolan and Cribb, Reference Nolan and Cribb2005). Given that cryptic species vary in key species-specific traits, and that trematodes have been found to play keystone roles in intertidal ecosystems (Mouritsen and Poulin, Reference Mouritsen and Poulin2002) by mediating biodiversity (Mouritsen and Poulin, Reference Mouritsen and Poulin2005) and productivity (Wood et al. Reference Wood, Byers, Cottingham, Altman, Donahue and Blakeslee2007) through altered host phenotype, it is important to explore their true diversity in order to understand the mediating role they can play in intertidal ecosystem functioning.
The New Zealand cockle, Austrovenus stutchburyi, serves as the second intermediate host for 2 species of trematodes that encyst in its foot. These two species, Curtuteria australis (Allison, Reference Allison1979) and Acanthoparyphium sp. of Martorelli et al. (Reference Martorelli, Poulin and Mouritsen2006), are both from the subfamily Himasthlinae and of the family Echinostomatidae. Due to similarity in their abundance levels and the identical ecological roles they appear to play in the soft-sediment intertidal ecosystem, they are considered to be ecological equivalents (Babirat et al. Reference Babirat, Mouritsen and Poulin2004). These two trematodes manipulate their cockle host by impairing its ability to burrow into the sediment (Thomas and Poulin, Reference Thomas and Poulin1998; Mouritsen, Reference Mouritsen2002). This not only facilitates their transmission to shorebird definitive hosts, but it also results in marked changes to the structure and diversity of the whole intertidal benthic community (Thomas et al. Reference Thomas, Renaud, de Meeüs and Poulin1998a; Mouritsen and Poulin, Reference Mouritsen and Poulin2005). While they utilize the same second intermediate host and share a similar niche within that host, C. australis and Acanthoparyphium sp. have different first intermediate hosts: the former uses the mud whelk, Cominella glandiformis (see Allison, Reference Allison1979), and the latter the mud snail Zeacumantus subcarinatus (see Martorelli et al. Reference Martorelli, Poulin and Mouritsen2006).
Although the morphologies of both species have been described, here we use phylogenetic analyses of nuclear and mitochondrial genes to reveal that 2 or more genetically distinct lineages exist within both Acanthoparyphium sp. and C. australis. In addition, we uncover a distinct small-scale spatial segregation among the clades within each genus that is totally unexpected given the dispersal potential of avian definitive hosts.
MATERIALS AND METHODS
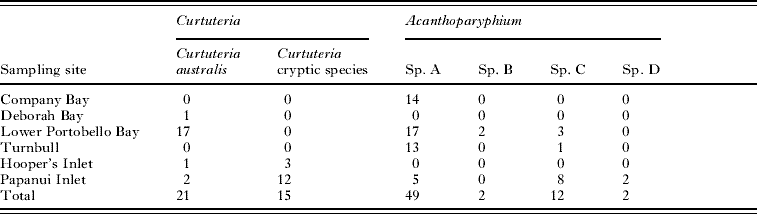
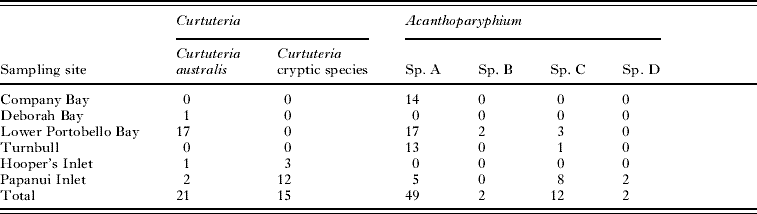
The parasites for this study were collected over the course of 3 years (Table 1). All hosts and parasites were collected from Otago Harbour and the nearby inlets along Otago Peninsula, South Island, New Zealand. Mud snails, Zeacumantus subcarinatus, were collected from Company Bay, Lower Portobello Bay, Turnbull Bay, and Papanui Inlet, while mud whelks, Cominella glandiformis, were collected from Deborah Bay, Lower Portobello Bay, Hooper's Inlet, and Papanui Inlet. Cockles, Austrovenus stutchburyi, were also collected from Company Bay and Lower Portobello Bay (see Fig. 1). The bivalve Macomona liliana was also collected from Company Bay, as it is also known to harbour foot-encysting echinostome metacercariae (Leung and Poulin, Reference Leung and Poulin2008). The animals were all brought back alive to the laboratory and dissected for parasites; Acanthoparyphium sp. rediae from Z. subcarinatus, Curtuteria australis rediae from C. glandiformis, and the metacercariae of both species from A. stutchburyi and M. liliana.

Fig. 1. Map of the study area showing sites where infected hosts were collected. The collection sites are: CB, Company Bay; TB, Turnbull Bay; DB, Deborah Bay; LPB, Lower Portobello Bay; HI, Hooper's Inlet; PNI, Papanui Inlet.
Table 1. Sampling site and number of Curtuteria spp. and Acanthoparyphium spp. samples
Each redia or metacercaria was carefully isolated from host tissue and transferred into a Petri dish containing 0·22 μm-filtered water. They were then transferred into another Petri dish containing filtered water as before. This procedure rinsed away any residual host material. The parasites were then placed individually into a 1·5 ml Eppendorf tube for DNA extraction. DNA was extracted in 500 μl of 5% chelex containing 0·1 mg/ml proteinase K, incubated at 60°C for 4 h and boiled at 100°C for 8 min.
The mitochondrial 16S gene of all individual rediae/metacercariae sampled was sequenced and the internal transcribed spacer 1 (ITS1) nuclear gene of a subset of representative samples from each of the separate clades was also sequenced subsequently. The 16S gene was amplified using platyhelminth-specific 16S primers platy.16Sar [5′-ATCTGTTT(A/C)T(C/T)AAAAACAT-3′] and platy.16Sbr [5′-CCAATCTTAACTCAACTCATAT-3′] as designed by Donald et al. (Reference Donald, Kennedy, Poulin and Spencer2004). The optimum cycling parameters for these primers included an initial denaturation step of 95°C (2 min), followed by 40 cycles of 95°C (30 s), 48°C (40 s) and 72°C (1 min), followed by a final extension phase at 72°C (10 min). The ITS1 gene was amplified using 2 primers described by Bowles and McManus (Reference Bowles and McManus1993), BD1 [5′-GTCGTAACAAGGTTTCCGTA-3′] and 4S [5′-TCTAGATGCGTTCGAA(G/A)TGTCGATG-3′]. The optimal cycling parameters for these primers included an initial denaturation of 94°C (2 min), followed by 40 cycles of 94°C (30 s), 52°C (1 min) and 71°C (1 min), followed by a final extension phase at 71°C (10 min). The PCR products were purified using Purelink™ PCR Purification kits (Invitrogen) and sequenced using the BigDye Terminator Cycle Sequencing Kit (Applied Biosystems), and resolved with an ABI PRISM 3730 Genetic Analyser (Applied Biosystems). Sequences were aligned using ClustalW in MEGA version 3.1 genetic analysis programme (Kumar et al. Reference Kumar, Tamura and Nei2004). Pairwise comparisons of sequences were conducted using Kimura's two parameter model (Kimura, Reference Kimura1980) also in MEGA.
Phylogenetic analyses were performed with PAUP* version 4b10 (Swofford, Reference Swofford2002) for maximum likelihood (ML) searches and bootstrap values (Felsenstein, Reference Felsenstein1985). To reduce computation time, for the Acanthoparyphium tree, only 2 sequence replicates from each haplotype were retained for the phylogenetic analyses. For the maximum likelihood analysis, the optimal model was identified using MODELTEST (Posada and Crandall, Reference Posada and Crandall1998), and for bootstrap analysis, 1000 replicates with 5 random addition sequences per replicate with NNI branch swapping were used.
Bayesian analyses were performed using MrBayes v3.1.2 (Huelsenbeck and Ronquist, Reference Huelsenbeck and Ronquist2001) with the following settings: the ML employed 2 substitution types (‘nst=2’). Rate variation across sites was modelled using a gamma distribution. The Markov-chain Monte-Carlo search was run with 4 chains for 500 000 generations, with trees being sampled every 100 generations (the first 1000 trees, i.e. 100 000 generations, were discarded as ‘burnin’).
For rooting of the Curtuteria phylogenetic tree, 2 representative sequences obtained for Acanthoparyphium were assigned as outgroup taxa. In turn, for the Acanthoparyphium phylogenetic tree, 2 representative sequences obtained for Curtuteria were assigned as outgroup taxa. The 16S and ITS1 sequence for Fasciola hepatica was also added as an outgroup taxon for each of the trees. F. hepatica was selected because of the availability of its 16S and ITS1 sequence in the GenBank database, and higher level phylogenetic analyses have found Fasciolidae to be a sister group to the Echinostomatidae (see Olson et al. Reference Olson, Cribb, Tkach, Bray and Littlewood2003).
The Fisher's exact test was used to compare the relative incidence of the different clades among infected snails between sites located in the harbour and those from the inlets.
RESULTS
A total of 36 Curtuteria-infected C. glandiformis were collected; 1 from Deborah Bay, 17 from Lower Portobello Bay, 4 from Hooper's Inlet and 14 from Papanui Inlet (Table 1). The 16S gene of 12 Curtuteria metacercariae (identified based on the presence of 31 collar spines) dissected from A. stutchburyi collected from Lower Portobello Bay was sequenced. A total of 78 Acanthoparyphium-infected Z. subcarinatus were collected; 22 from Lower Portobello Bay, 14 from Company Bay, 14 from Turnbull Bay, and 28 from Papanui Inlet (Table 1). The 16S gene of 11 Acanthoparyphium metacercariae (identified based on the presence of 23 collar spines) dissected from A. stutchburyi and M. liliana collected from Lower Portobello Bay and Company Bay was sequenced. PCR amplification of 16S yielded 427 bp of readable sequence (GenBank Accession numbers FJ396045–FJ396142), for Acanthoparyphium and Curtuteria, and ITS1 yielded 555 bp of readable sequence (GenBank Accession numbers FJ396143– FJ396164) for both Acanthoparyphium and Curtuteria samples.
Parasites identified as Curtuteria were found to belong to 2 different clades (Table 1 and Figs 2 and 4), and Acanthoparyphium was found to be composed of 4 highly divergent clades (Table 1 and Figs 3 and 5). The trees produced by ML and Bayesian analyses gave statistical support for the existence of distinct genetic species, with the topology of trees produced with data from the 16S gene (Figs 2 and 3) concordant with the trees produced using the ITS1 gene (Figs 4 and 5).

Fig. 2. Phylogenetic relationships of the Curtuteria spp. samples isolated from Cominella glandiformis inferred from 16S sequences. The codes correspond to the collection sites shown in Fig. 1. The first number associated with each node represents the ML bootstrap value, followed by the Bayesian posterior probabilities.

Fig. 3. Phylogenetic relationships of the Acanthoparyphium spp. samples isolated from Zeacumantus subcarinatus inferred from 16S sequences. The codes correspond to the collection sites shown in Fig. 1. The first number associated with each node represents the ML bootstrap value, followed by the Bayesian posterior probabilities.

Fig. 4. Phylogenetic relationships of the Curtuteria spp. samples isolated from Cominella glandiformis inferred from ITS1 sequences. The codes correspond to the collection sites shown in Fig. 1. The first number associated with each node represents the ML bootstrap value, followed by the Bayesian posterior probabilities.

Fig. 5. Phylogenetic relationships of the Acanthoparyphium spp. samples isolated from Zeacumantus subcarinatus inferred from ITS1 sequences. The codes correspond to the collection sites shown in Fig. 1. The first number associated with each node represents the ML bootstrap value, followed by the Bayesian posterior probabilities.
The levels of 16S sequence divergence between cryptic species were high, ranging from 13·1 to 13·7% for the 2 Curtuteria species (Table 2) and from 5·8 to 11·1% between the cryptic Acanthoparyphium species (Table 3). The slower-evolving ITS1 sequence showed considerably less divergence, with only 1·8% divergence between the 2 Curtuteria species (Table 2) and 0·6–1·6% divergence between the Acanthoparyphium species (Table 3).
Table 2. Range of percentage sequence divergence at 16S and ITS1 within and between the Curtuteria spp.

Table 3. Range of percentage sequence divergence at 16S and ITS1 within and between the Acanthoparyphium spp.

The level of within clade divergence for 16S sequences was between 0·0 and 0·5% for the Curtuteria clades and between 0·0 and 1·1% for the Acanthoparyphium clades. For the ITS1 sequences, the level of divergence within each of the Curtuteria clades was 0·0% (Table 2), whereas within each Acanthoparyphium clades, the level of sequence divergence was 0·0–0·2% (Table 3).
The 16S sequences of 12 Curtuteria spp. metacercariae (identified based on the presence of 31 collar spines) dissected from cockles collected from Lower Portobello Bay were found to match the sequences of the ‘harbour clade’ of Curtuteria (i.e. C. australis) with less than 0·5% divergence. The 16S sequences of the 11 Acanthoparyphium metacercariae (identified based on the presence of 23 collar spines) dissected from A. stutchburyi and M. liliana collected from Company Bay and Lower Portobello Bay were found to match the sequences of Acanthoparyphium sp. A, with less than 0·6% divergence.
All Curtuteria sequenced from the harbour belong to the same monophyletic clade, while 15 out of 18 of the Curtuteria sequenced from the inlets were of a separate clade (Table 1 and Fig. 6A). Of the harbour sample, Acanthoparyphium sp. A accounted for 44 out of the 50 samples sequenced, whereas 2 were from sp. B and 4 from sp. C. In contrast, of the inlet sample, 9 out of 28 samples were of sp. A, 14 were of sp. C, and 5 were of sp. D (Table 1 and Fig. 6B). Acanthoparyphium sp. B was absent from the inlet sample while sp. D was absent from the harbour sample. This segregation pattern was statistically significant for both genera (Fisher's exact test, both P<0·0001).

Fig. 6. The relative incidence of the Curtuteria spp. (A) and the Acanthoparyphium spp. (B) among infected snails with respect to their site of origin.
DISCUSSION
This study has uncovered a previously unsuspected level of diversity in the trematodes of Otago Harbour in the form of genetically distinct clades within otherwise morphologically identical or very similar groups of individuals. According to Vilas et al. (Reference Vilas, Criscione and Blouin2005), the maximum intraspecific divergence for trematodes in mitochondrial DNA sequences ranges from 0·3 to 2·2%. The levels of variation seen in this study, combined with phylogenetic support for the clades, indicate that the clades represent genetically distinct species. The level of ITS1 sequence divergence between the different clades/species of this study are consistent with the level of interspecific variations expected for the family Echinostomatidae (reviewed by Nolan and Cribb, Reference Nolan and Cribb2005).
The Curtuteria clade that encompasses all samples from whelks collected from Deborah Bay and Lower Portobello Bay, plus a few of the individuals from Hooper's Inlet and Papanui Inlet, is most likely Curtuteria australis described by Allison (Reference Allison1979). This conclusion is based on the 16S sequences of metacercariae with 31 collar spines dissected from cockles collected from Lower Portobello Bay, which closely matched those obtained from rediae dissected from whelks and previously assigned to C. australis (see Babirat et al. Reference Babirat, Mouritsen and Poulin2004). The Acanthoparyphium metacercariae with 23 collar spines dissected from bivalves collected from Company Bay and Lower Portobello Bay were identical to sequences from Acanthoparyphium sp. A. This indicates that the clade that we have named sp. A in the present study most likely corresponds to Acanthoparyphium sp. described by Martorelli et al. (Reference Martorelli, Poulin and Mouritsen2006) from metacercariae.
The discovery of cryptic species of Acanthoparyphium adds to the growing list of trematodes known to use the mudsnail Z. subcarinatus as a first intermediate host. This snail is already known to be host to at least 6 described species of trematodes (Martorelli et al. Reference Martorelli, Fredensborg, Mouritsen and Poulin2004, Reference Martorelli, Poulin and Mouritsen2006, Reference Martorelli, Fredensborg, Leung and Poulin2008). The results of this study not only shed new light on the diversity of trematodes infecting Z. subcarinatus, but also add weight to the possibility that many previously described species of trematodes found in other intertidal ecosystems in fact consist of cryptic species complexes (e.g. Miura et al. Reference Miura, Kuris, Torchin, Hechinger, Dunham and Chiba2005).
The life cycles of the cryptic echinostome species found in this study are yet to be elucidated, though they are likely similar to those of C. australis and Acanthoparyphium sp. (sp. A). One way of elucidating their life cycles would involve prospecting for parasites from potential second intermediate hosts collected from the inlets where the cryptic clades appear to be more common. While the metacercariae of Curtuteria spp. are only known to infect bivalves, those of Acanthoparyphium can infect prosobranch and pulmonate gastropods, as well as polychaetes, in addition to bivalves (Kostadinova, Reference Kostadinova, Jones, Bray and Gibson2005). The cryptic species found in this study may also infect the cockle A. stutchburyi, but encyst in parts of the cockle's body other than its foot where Acanthoparyphium sp. A and C. australis co-occur. Different species of Himasthla, which infect the European cockle Cerastoderma edule, show different host tissue preferences for encystment (Wegeberg et al. Reference Wegeberg, de Montaudouin and Jensen1999; de Montaudouin et al. Reference De Montaudouin, Jensen, Desclaux, Wegeberg and Sajus2005); therefore, the presence of the cryptic species in A. stutchburyi may simply have been overlooked. However, all echinostome metacercariae found in cockles in the Otago area show a specific preference for the tip of the host's foot (Mouritsen Reference Mouritsen2002; Babirat et al. Reference Babirat, Mouritsen and Poulin2004) and rarely encyst elsewhere. Site segregation within the cockle host between these cryptic species may occur at a much finer scale, i.e. within different subsections of the foot. Mouritsen (Reference Mouritsen2002) found that the foot-encysting echinostomes in cockles alter the host's burrowing behaviour by simple mechanical obstruction, and although metacercariae occur throughout the cockle's foot, only those encysted at the tip of the foot actually impair host burrowing. In addition, the manipulation of cockle behaviour comes at considerable risk in the form of predation by non-host fish predators that crop the tip of the cockle's foot (Mouritsen and Poulin, Reference Mouritsen and Poulin2003). While previous findings suggest that both Acanthoparyphium sp. and C. australis contribute equally to host manipulation by preferentially encysting at the tip of the cockle's foot (Babirat et al. Reference Babirat, Mouritsen and Poulin2004), in light of the present results, it is possible that niche partitioning is occurring between the cryptic species of echinostomes. Certain cryptic species may be ‘hitch-hikers’ that do not manipulate host behaviour, but associate themselves with parasites that do alter host phenotype (Thomas et al. Reference Thomas, Renaud and Poulin1998b). By encysting away from the tip, these metacercariae can exploit the enhanced transmission rate induced by manipulative clades, but without incurring the fish predation risk associated with host manipulation.
Apart from the discovery of 6 cryptic species among trematodes previously described as just 2 species, our most striking finding is the extremely localized pattern of distribution of these species. For example, while the cryptic Curtuteria species is 5 times more common than C. australis in sheltered inlets, it is either absent or very rare in the Otago Harbour. A similar small-scale spatial segregation pattern is also apparent among the Acanthoparyphium species. This is particularly perplexing considering that the definitive hosts of these trematodes are birds (Kostadinova, Reference Kostadinova, Jones, Bray and Gibson2005) that can disperse infective stages across wide geographical areas (Criscione and Blouin, Reference Criscione and Blouin2004; Miura et al. Reference Miura, Torchin, Kuris, Hechinger and Chiba2006; Keeney et al. Reference Keeney, Bryan-Walker, King and Poulin2008). The marked segregation of these cryptic species on a scale of a few kilometres therefore warrants a discussion of potential explanations.
First, and most parsimoniously, the patterns seen in Fig. 6 could simply be the result of a sampling artefact, especially given the small sample size for the Curtuteria species. However, this pattern is repeated in Acanthoparyphium spp. for which a larger number of individuals were sequenced. While a larger sample would further clarify the distribution pattern of these cryptic species, regardless of sample size, the fact that 2 different genera that utilize 2 different species of gastropod as first intermediate hosts display a similarly uneven distribution makes it difficult to dismiss as a mere by-product of inadequate sampling.
Secondly, the distribution pattern may be the consequence of local adaptation: either by the parasite towards a particular host population, or by a host population against a particular parasite. While trematodes are highly specific for their first intermediate host (Gibson and Bray, Reference Gibson and Bray1994), mainly because of molluscan defence responses (Sapp and Loker, Reference Sapp and Loker2000; Bayne et al. Reference Bayne, Hahn and Bender2001), this specificity does not extend to specialization on a particular host population. Local adaptation on the part of the parasite seems even less likely given that avian definitive hosts can disperse the larval stages over great distances, resulting in genetically homogeneous parasite populations (Criscione and Blouin, Reference Criscione and Blouin2004; Keeney et al. Reference Keeney, Bryan-Walker, King and Poulin2008). Local adaptation by the gastropod host appears more likely. Because both Z. subcarinatus and C. glandiformis produce crawling larvae without planktonic veliger stages (Pilkington, Reference Pilkington1974), gastropods within either the bays or inlets could represent isolated populations that vary in their susceptibility to infection by different trematode species. Lively and Jokela (Reference Lively and Jokela1996) have reported that freshwater snails of the same species sampled along a depth gradient in a single lake varied in their susceptibility to infection by the same trematode species. However, while populations of Z. subcarinatus from bays with high parasite prevalence exhibit life-history adaptations that minimize the negative impact of trematode infection, such as maturing at a smaller size, no variation was found in the susceptibility of snails from these different populations to trematode infections (Fredensborg and Poulin, Reference Fredensborg and Poulin2006).
Thirdly, differences in environmental conditions between the harbour and inlets could give certain species a competitive advantage over others in terms of successfully infecting the appropriate molluscan host. Several habitat features can contribute to spatial heterogeneity in species composition of snail-trematode communities (Williams and Esch, Reference Williams and Esch1991; Koprivnikar et al. Reference Koprivnikar, Baker and Forbes2007). Both biotic and abiotic factors affect the survival of free-living stages of endohelminths, including trematodes (Pietrock and Marcogliese, Reference Pietrock and Marcogliese2003; Thieltges et al. Reference Thieltges, Jensen and Poulin2008). However, these environmental conditions generally have broad-spectrum effects with very little specificity, certainly not enough to target one species of a genus but not another. It is difficult to pinpoint potential environmental factors that differ between the harbour and inlets and that could only affect a particular species while not affecting congeners, let alone having a similar impact on another genus. Thus, differences in local factors, both abiotic and biotic, are unsatisfactory as potential explanations for the observed spatial segregation of cryptic species.
Fourthly, while the environment may not be directly responsible for the segregated distribution of these species, because they use the definitive host as their main means of dispersal, environmental factors may still shape distribution patterns by influencing the spatial distribution of shorebirds. There is a positive association between habitat usage by shorebirds and the recruitment rate of trematodes in local snail populations (Smith, Reference Smith2001; Hechinger and Lafferty, Reference Hechinger and Lafferty2005; Fredensborg et al. Reference Fredensborg, Mouritsen and Poulin2006; Whitney et al. Reference Whitney, Hechinger, Kuris and Lafferty2007; Byers et al. Reference Byers, Blakeslee, Linder, Cooper and Maguire2008). Thus the presence of birds at a particular site could modulate the subsequent distribution of different trematode species. Habitat choices of shorebirds when selecting a site for feeding, breeding, or roosting are influenced by a number of factors including substrate characteristics (Mouritsen and Jensen, Reference Mouritsen and Jensen1992; Finn et al. Reference Finn, Catterall and Driscoll2007) and prey availability (Ribeiro et al. Reference Ribeiro, Iribarne, Navarro and Jaureguy2004; Jing et al. Reference Jing, Ma, Li, Li and Chen2007), which in turn can be affected by the presence of bioturbators such as burrowing crustaceans (Iribarne et al. Reference Iribarne, Bruschetti, Escapa, Bava, Botto, Gutierrez, Palomo, Delhey, Petracci and Gagliardini2005). Divergent site preferences by different shorebird species or individuals infected by different parasite species could possibly maintain the localized distribution seen in the two echinostome species complexes.
None of the above is a very likely or convincing explanation, however. The limited distance between Otago Harbour and the surrounding inlets make it difficult to attribute the observed distribution pattern to bird movement. The somewhat unexpected patterns found here present a tantalizing glimpse into a biological puzzle that warrant further and more intensive investigation. Future approaches should include: larger sample sizes, elucidation of the life cycles of all cryptic species, development of a faster and cheaper method of identifying the cryptic species (such as RFLP; Donald et al. Reference Donald, Sijnja and Spencer2007) since cockles commonly harbour hundreds of echinostome metacercariae (Leung and Poulin, Reference Leung and Poulin2007), and possibly conducting bird surveys to ascertain if host movements can generate the distribution pattern found for the cryptic species. Assuming that the present findings are valid, we may have to reconsider some of the accepted but largely untested tenets regarding the trematode life cycle with respect to host specificity and dispersal potential.
The authors would like to thank Kim Bryan-Walker and Ian Saldanha for assistance with field collection and dissection, and three anonymous referees for their comments.