


INTRODUCTION
Filarial nematodes of the genus Onchocerca have a worldwide distribution, infecting humans, ruminants, camels, horses, suids, and canids, with effects ranging from relatively benign to severely debilitating (Anderson, Reference Anderson2000; Boatin and Richards, Reference Boatin and Richards2006; Sreter and Szell, Reference Sreter and Szell2008). Thus, this group of parasites evidently has considerable evolutionary potential to shift among mammalian host species. Furthermore, several species of Onchocerca occur in 2 or more disjunct biogeographical realms, including species that infect humans (O. volvulus, which causes human onchocerciasis), cattle (O. gutturosa and O. lienalis), and horses (O. cervicalis, and O. reticulata) (Anderson, Reference Anderson2000; Basanez et al. Reference Basanez, Pion, Churcher, Breitling, Little and Boussinesq2006). Such widespread distributions suggest that competent intermediate hosts (blackfly species: Simuliidae) are geographically broadly available and that the ability of some Onchocerca species to switch between intermediate hosts during range expansions may contribute to the weak biogeographical signal in the Onchocerca phylogeny (Krueger et al. Reference Krueger, Fischer and Morales-Hojas2007).
The combination of an evolutionary history of host shifts and widespread occurrence of competent vectors suggest that the genus Onchocerca has considerable potential to colonize currently uninfected host populations. Given the possible evolutionary lability of Onchocerca infections, it is desirable to survey the diversity of extant Onchocerca species and to formally test their capacity to shift to a new vertebrate host species.
A considerable fraction of extant Onchocerca species are associated with ungulates, in particular cervids. For instance, Cervus elaphus (red deer) in Eurasia can be infected with O. flexuosa, O. garmsi, O. jakutensis (=O. tubingensis) and O. skrjabini (=O. tarsicola) (Bain and Schulz-Key, Reference Bain and Schulz-Key1974, Reference Bain and Schulz-Key1976; Demiaszkiewicz, Reference Demiaszkiewicz1992, Reference Demiaszkiewicz1993). To date, however, only 1 species, O. cervipedis, has been found to infect North American cervids (Hibler, Reference Hibler1965; Wehr and Dikmans, Reference Wehr and Dikmans1935; Weinmann et al. Reference Weinmann, Anderson, Longhurst and Connolly1973; Bain and Schulz-Key, Reference Bain and Schulz-Key1974; Low, Reference Low1976; Samuel et al. Reference Samuel, Barrett and Lynch1976). Here we report the discovery of a second cervid-infecting species of Onchocerca in North America. We use DNA sequence data to reconstruct the phylogenetic history of Onchocerca, including inferences of host usage patterns throughout the evolutionary history of the genus.
MATERIALS AND METHODS
Sample collection
To obtain Onchocerca microfilariae from white-tailed deer (Odocoileus virginianus), 3 contractors involved with removal of road-killed deer in upstate New York were coordinated to collect both pinnae of the ears from recently killed deer. Samples were collected from the surrounding areas of Rochester (43·11°N, 77·63°W), Campbell (42·23°N, 77·20°W), and Glens Falls, New York (43·31N, 73·65°W). Eighty-four deer were collected in the counties surrounding Campbell, while 2 came from Rochester and 13 from Glens Falls. The samples were either shipped to the University of Rochester on ice or stored in a 4 °C refrigerator for weekly collection. Adults were not isolated from New York deer, and therefore morphology could not be used for identification. The goals of the study, however, were to explore the diversity of Onchocerca in North America and evolutionary patterns of host shifts in the genus, and adults were therefore not necessary.
Microfilariae were extracted from deer ears using a procedure developed for extracting Onchocerca species from cattle (Neary et al. Reference Neary, Trees, Ekale, Tanya, Hetzel and Makepeace2010). The procedure outlined by Neary et al. (Reference Neary, Trees, Ekale, Tanya, Hetzel and Makepeace2010) was modified only by the addition of 200 U/ml penicillin, 200 μg/ml streptomycin and 0·5 μg/ml amphotericin B to the RPMI 1640 cell culture medium that was used to extract microfilariae.
As part of a separate study, one of the authors (Verocai) obtained isolates of O. cervipedis from 2 moose (Alces alces) from Moose Tabasco Lake (65·28°N, 131·12°W) and Moose Kelly Lake (65·44°N, 126·18°W), both in the Northwest Territories, Canada. For the analyses presented here, adult females were obtained from one moose, and microfilariae and an adult female from a second moose. Adult worms were collected from nodules in the subcutaneous tissues of the metatarsals of both moose. The nematodes were morphologically consistent with descriptions of O. cervipedis, as was their location in the host (Verocai et al. in the Press).
Molecular genetic techniques
To determine the phylogenetic position of the microfilariae obtained from white-tailed deer and O. cervipedis from moose, 2 mitochondrial rRNA genes (12S and 16S) and 1 mitochondrial protein–coding gene (mitochondrially encoded NADH dehydrogenase 5, hereafter ND5) were sequenced. These genes were chosen because they have been used for previous Onchocerca phylogenetics, which allowed us to use publicly available sequences to include in our phylogeny (Morales-Hojas et al. Reference Morales-Hojas, Cheke and Post2006; Krueger et al. Reference Krueger, Fischer and Morales-Hojas2007). Onchocerca DNA was extracted from 50 or more pooled microfilariae isolated from 5 different deer with the PureGene DNA purification kit (Qiagen, Valencia, CA). The 12SOv, 16SOv, and ND5Ov primers (Morales-Hojas et al. Reference Morales-Hojas, Cheke and Post2006) were used, along with a touchdown PCR protocol that went from 58 °C for the initial annealing temperature to 55 °C for the final annealing temperature. The resulting PCR products were then cloned using a TOPO-TA cloning kit (Invitrogen, Carlsbad, CA, USA). Five colonies from these cloning reactions were selected and the M13 primers and a 50 °C annealing temperature were used to perform PCR. Big-Dye reagents were then used and the resulting products were sequenced in both directions on an ABI Prism 3700 (Applied Biosystems, Carlsbad, CA, USA).
DNA was extracted from 1 adult female O. cervipedis from each moose using the DNeasy purification kit (Qiagen, Valencia, CA, USA). For PCR and DNA sequencing, the same protocol as described above was used , but it was found that high quality sequence could be obtained without cloning. Therefore PCR products were directly sequenced in both directions.
Wolbachia is a group of widespread endosymbiotic bacteria that are obligate symbionts in important filarial parasites of humans and animals (Bandi et al. Reference Bandi, Trees and Brattig2001). To determine whether our Onchocerca samples associate with Wolbachia, our DNA extractions were PCR screened using multiple Wolbachia primer sets. To characterize Wolbachia, the pubMLST primer sets were used to amplify 5 genes used for multilocus sequence typing (MLST) of Wolbachia: gatB, coxA, hcpA, ftsZ and fbpA (http://pubmlst.org/wolbachia, accessed 5/2/2012, Jolley et al. Reference Jolley, Chan and Maiden2004). Additionally, the pubMLST wsp primers were used, along with wspec (Werren and Windsor, Reference Werren and Windsor2000) and 16SWolb (Casiraghi et al. Reference Casiraghi, Anderson, Bandi, Bazzocchi and Genchi2001), both of which amplify Wolbachia 16S. For all Wolbachia PCR reactions, a touchdown protocol with 10 cycles was used, ramping the annealing temperature down 1 degree per cycle from 58 °C-48 °C, followed by 30 cycles with a 48 °C annealing temperature. The resulting positive PCR products were directly sequenced in both directions.
The DNA sequences from this study were archived on the genetic sequence database at the National Center for Biotechnology Information (NCBI) (GenBank IDs: JX075199-JX075238), and additionally, novel Wolbachia alleles and isolate information (isolates 505 and 506) were deposited on the pubMLST website.
DNA sequence alignment
For phylogenetic reconstructions, all available Onchocerca sequences for the 16S (33 sequences from 13 species), 12S (56 sequences from 20 species) and ND5 (39 sequences from 16 species) genes, as of 13 July 2011 were downloaded from NCBI GenBank (for accession numbers and references, see Table S1, online version only). For use as outgroups, sequences from other filariid nematodes were incorporated, including Brugia malayi, Chandlerella quiscali, Dirofilaria immitis, Loa loa, Litomosoides carinii, and Setaria digitata. Four different datasets were compiled: an alignment with all 3 genes concatenated for each of our samples and for each publicly available sequence where the metadata associated with the separate genes allowed us to determine that the sequences came from the same sample (S1, online version only), and 3 separate alignments for 12S (S2, online version only), 16S (S3, online version only), and ND5 (S4, online version only), representing all of the available sequences for each gene along with our sequences for each gene.
Because cloned PCR products were used for DNA sequencing, it was assumed that sequences with single nucleotide polymorphisms that showed up in only 1 of 5 cloned sequences were the result of PCR error and thus these sequences were discarded. After accounting for PCR error, it was determined that 2 of the deer in our samples were co-infected with nematodes carrying distinct mtDNA haplotypes at the 12S and ND5 loci (161 and 347). To concatenate sequences from co-infected deer, the sequences were first combined in every possible configuration, and preliminary maximum likelihood analyses were run. Combinations of sequences were then selected from the doubly infected deer that closely matched combinations of sequences found in singly infected deer. It was therefore assumed that worm genotypes found in singly infected deer also occur in doubly infected deer.
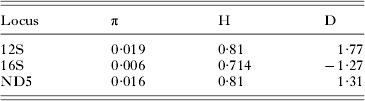
Muscle (Edgar, Reference Edgar2004) was used to align homologous sites in each alignment and the alignments were further refined by eye in the program Mesquite (Maddison and Maddison, Reference Maddison and Maddison2010). It was found that sections of our single gene 12S alignment (which included more sequences than the concatenated alignment) were not easily assigned to homologous positions, and therefore these sites were excluded from subsequent analyses (see S2 for nexus file identifying excluded sites). Table 1 lists the simple sequence statistics for each of these alignments, while Table 2 lists the nucleotide diversity, haplotype diversity, and Tajima's D of the sequences from microfilariae associated with New York deer, as calculated with DNAsp (Rozas et al. Reference Rozas, Sanchez-DelBarrio, Messeguer and Rozas2003).
Table 1. Simple sequence statistics for four Onchocerca DNA sequence alignments
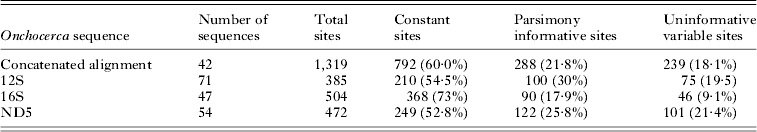
Table 2. Nucleotide diversity (π), haplotype diversity (H), and Tajima's D (D) of the New York deer-associated Onchocerca species
Onchocerca phylogenetic analyses
Two different methods were used for phylogenetic reconstructions: approximate maximum likelihood, as implemented in the program GARLI 2.0 (Zwickl, Reference Zwickl2006), and Bayesian phylogenetics, as implemented in MrBayes v.3.1.5 (Ronquist and Huelsenbeck, Reference Ronquist and Huelsenbeck2003). To perform approximate maximum likelihood analyses, the most likely model of sequence evolution was first determined using the program Modeltest 3.7 (Posada and Crandall, Reference Posada and Crandall1998) and PAUP* 4b10 (Swofford, Reference Swofford2002). Several different partitioning schemes were tested for the concatenated dataset: (1) a single model for the entire dataset; (2) separate models for each gene; (3) separate models for 12S, 16S, the 1st and 2nd codon positions for ND5 (treated as one category), and the 3rd codon positions for ND5; and (4) separate partitions for each rDNA gene and each codon position for ND5. Four approximate maximum likelihood search replicates were then run for each of the 4 partitioning schemes in GARLI, using the most likely model of evolution for each partition, as selected by Modeltest, but allowing parameters to be estimated during the run. To determine the best partitioning scheme for phylogenetic reconstruction, the –ln likelihood of the best scoring search replicate for each scheme and the number of parameters in its embedded models were used to calculate an AIC score. The scheme with the lowest scoring AIC (15538·19) had separate models for 12S (GTR + Γ + I), 16S (GTR + Γ + Ι), and each codon position for ND5 (GTR + Γ, K81uf + Γ, and K81uf + Γ for 1st, 2nd and 3rd codon positions respectively). To assess support for the tree topology, 100 bootstrap pseudoreplicates were run. Likelihood searches were additionally performed on each gene separately, using the most likely model of evolution as selected above for each respective gene. For the single gene analyses, 10 replicate searches for the best tree and 100 bootstrap pseudoreplicates were run.
To determine the best fitting model of evolution for the Bayesian phylogenetic runs, MrModeltest 2.3 (Nylander, Reference Nylander2004) was used. Analyses using 3 different partitioning schemes in MrBayes v.3.1.5 (Ronquist and Huelsenbeck, Reference Ronquist and Huelsenbeck2003) were run: (1) a single model of evolution underlying all 3 genes; (2) separate models of evolution for each gene; and (3) separate models of evolution for the rRNA genes and for each codon position in ND5. For each analysis, 2 independent runs with 4 Markov chains each were run for 20 000 000 generations. The chain was sampled every 1000 generations and the first 25% of the generations were discarded as burn-in. Analysis of the resulting parameter files using tracer V1·5 (Rambaut and Drummond, Reference Rambaut and Drummond2009) confirmed that the number of generations discarded as burn-in was appropriate. The best run, as determined by Bayes factors computed from the harmonic means of the likelihood values from each run (Kass and Raftery, Reference Kass and Raftery1995), utilized separate models for 12S (GTR + Γ + I), 16S (GTR + Γ + Ι) and codon positions in ND5 (separate GTR + Γ models) for first, second and third codons. To confirm that the runs had converged, the average standard deviation of split frequencies was examined, and the split and compare commands in AWTY were executed (Wilgenbush et al. Reference Wilgenbush, Warren and Swofford2004).
Wolbachia phylogenetic analyses
To explore the phylogenetic history of Wolbachia associated with Onchocerca, 16S sequences were obtained from 3 microfilarial samples from deer and from 2 adult O. cervipedis samples from moose using the wspec primers (Werren and Windsor, Reference Werren and Windsor2000). Additionally, all of the 16S sequences from Onchocerca-associated Wolbachia that were available on GenBank as of 1 May 2012 were downloaded. For outgroup sequences, 5 Wolbachia sequences that had been isolated from Dirofilaria species were downloaded (see Table S2 for accession numbers and references, online version only). After aligning the sequences using Muscle (Edgar, Reference Edgar2004) in the program Mesquite (Maddison and Maddison, Reference Maddison and Maddison2010), it was found that our coverage of the 16S gene overlapped with 11 GenBank sequences, so our alignment was trimmed to include only these sites and sequences. This alignment lacked a strong phylogenetic signal; from a total of 460 sites, only 13 were parsimony informative, while 19 were uninformative, variable characters.
Maximum likelihood analyses were conducted on our Wolbachia 16S alignment using GARLI 2.0 (Zwickl, Reference Zwickl2006). The most likely model of sequence evolution (K80+ Γ) was first determined using Modeltest 3.7 (Posada and Crandall, Reference Posada and Crandall1998) and PAUP* 4b10 (Swofford, Reference Swofford2002). Four search replicates for the most likely tree, and 100 bootstrap pseudoreplicates were run.
Ancestral state reconstructions
To reconstruct the host associations of ancestral nodes in our phylogenetic hypotheses, both Bayesian and maximum likelihood reconstruction methods were employed. First, the program BayesTraits v1.0 (Pagel et al. Reference Pagel, Meade and Barker2004) was used to analyse our three-gene dataset. To account for phylogenetic uncertainty in our trees, we randomly selected 10 000 post burn-in trees from our MrBayes analysis (5 000 from each run). Reversible-jump MCMC was used to allow the analysis to find the models that best fit our data (Pagel and Meade, Reference Pagel and Meade2006), utilizing an exponential distribution seeded on the interval of 0–30 from a uniform distribution. To allow for adequate exploration of the rate parameters, a ratedev setting of 15 was used. To reconstruct ancestral states, the MRCA command was used and 5 independent runs were conducted. Each analysis was run for 100 000 000 generations and the first 25 000 000 generations were discarded as burn-in.
To include a more complete sampling of species and subsequent host associations than permitted in our three-gene analysis, maximum likelihood ancestral state reconstruction was used on our single gene phylogenetic hypotheses in the program Mesquite (Maddison and Maddison, Reference Maddison and Maddison2010). The Markov k-state 1 parameter model of character evolution was used to determine the most likely ancestral character states, and the program was allowed to estimate model parameters based on the data.
RESULTS
Deer sampling and prevalence of Onchocerca
Twenty-three of 99 road-killed deer that were collected between 9 July 2010 and 29 November 2010 were infected with Onchocerca microfilariae. Neither of the 2 deer from the Rochester area was infected, while 18 of 84 (21·4%) and 5 of 13 (38·5%) deer from the Campbell and Glens Falls areas, respectively, were infected with Onchocerca. Microfilariae from 5 of these deer (3 from Glens Falls and 2 from Campbell) were used for our phylogenetic analyses.
Onchocerca phylogenetics
Several patterns arose from our phylogenetic analyses. First, the Onchocerca microfilariae isolated from deer in upstate New York formed a monophyletic group in our three-gene and ND5 analyses but not in our 12S and 16S rDNA analyses (Fig. 1 and Figs S1–S3, online version only). Because mtDNA is a non-recombining molecule, the concatenated sequence of all 3 mitochondrial genes (12S, 16S, and ND5) contains more phylogenetic information than do the individual genes and thus is more likely to yield a closer approximation to the true evolutionary history of this group. It should be noted, however, that several nodes in the three-gene phylogeny show low support values. In the 12S tree, 3 deer-associated microfilariae sequences clustered with O. lienalis, while 4 other deer isolated sequences clustered with O. jakutensis (Fig. S1, online version only). In the 16S analysis, 1 sample fell as sister to the rest of the New York samples, O. lienalis sequences, and O. jakutensis sequences (Fig. S2, online version only). All of the deeper nodes in this clade, however, had low support values. Microfilariae isolated from New York deer clustered with O. lienalis in the three-gene and ND5 trees (Fig. 1 and Fig. S3, online version only, respectively).

Fig. 1. Three-gene maximum likelihood tree of members of the genus Onchocerca. Values on top of nodes or next to nodes are posterior probability values/bootstrap support values from a 20 000 000 generation Bayesian analysis and 100 maximum likelihood bootstrap pseudoreplicates respectively. Host associations for extant taxa are shown on the right, with straight lines connecting host and Onchocerca clades. Host association for ancestral nodes, based on the results of our BayesTraits analyses, is shown on the left, with curved lines connecting hosts and ancestral nodes. The size of the host picture represents the posterior probability of host associations, which is also reported to the left of the host picture. Sequences generated for this study are 161_1, 161_2, 347_1, 347_2, DA2, DQ1, JL20, Onchocerca_cervipedis_1, and Onchocerca_cervipedis_2.
Next, Onchocerca isolated from deer in upstate New York did not cluster with O. cervipedis that were isolated from moose in Northwest Territories, Canada; in fact, these 2 species were found to be among the most distantly related within the genus Onchocerca. The O. cervipedis isolates clustered with O. armillata in all phylogenetic reconstructions except for the 16S tree (Fig. 1 and Figs S1–S3, online version only). Although O. cervipedis and O. armillata did not form a monophyletic group in the 16S phylogeny (Fig. S2, online version only), the nodes separating them showed low bootstrap support.
Our three-gene phylogenetic hypothesis suggested that the suid-associated O. ramachandrini is sister to the rest of the genus and that O. armillata and O. cervipedis are additionally sister to the clade that contains the bulk of Onchocerca diversity (Fig. 1). Our single-gene hypotheses further support these relationships, although another suid-associated species (O. dewittei) falls as sister in the 12S phylogeny (Fig. S1, online version only). It should also be noted that the deeper branches in the single-gene trees receive little bootstrap support (Figs S1–S3, online version only). Lastly, all of our phylogenetic analyses indicate that O. volvulus, O. ochengi, O. dukei and an unidentified species (‘siisa’) form a monophyletic group (Fig. 1 and Figs S1–S3, online version only).
Two of our phylogenetic results appeared to be anomalous, specifically the very long branch for 1 O. lienalis sample in the 12S phylogeny (Fig. S1, online version only), and the placement of Brugia malayi within Onchocerca in the ND5 phylogeny (Fig. S3, online version only). However, in the absence of any other reason to exclude these samples, we have chosen to include them in our phylogenetic analyses.
Wolbachia MLST and phylogenetics
Wolbachia 16S was amplified from 3 deer-associated Onchocerca samples and Wolbachia coxA from one deer-associated sample. Sequences were obtained from O. cervipedis-associated Wolbachia for 16S, coxA, gatB, fbpA, and ftsZ. We were unable to amplify hcpA, and were therefore unable to define a MLST strain type for either of our Onchocerca-associated Wolbachia strains. Wolbachia from O. cervipedis and deer-associated Onchocerca differed at both their 16S and coxA sequences. Both O. cervipedis samples had identical sequences at all loci. All 5 unique alleles that we queried against the pubMLST allele sequence database were novel and shared between 88·34% and 91·79% sequence identity with alleles in the database (http://pubmlst.org/wolbachia, accessed 2 May 2012).
Our Wolbachia 16S alignment exhibited little phylogenetic signal, but firmly placed Wolbachia from O. cervipedis and deer-associated Onchocerca sp. into the C-supergroup, along with Wolbachia from other Onchocerca and Dirofilaria species (Fig. 2). Additionally, our Wolbachia sequences formed a monophyletic group in the 16S phylogeny, albeit with low bootstrap support (Fig. 2).

Fig. 2. Maximum likelihood phylogeny of Wolbachia associated with Onchocerca, based on 16S rDNA. Values on branches represent bootstrap support from 100 pseudoreplicates, only values over 50% shown. Sequences generated for this study are 161, 347, JL20 (from deer associated Onchocerca), Onchocerca_cervipedis_1 and Onchocerca_cervipedis_2 (from O. cervipedis).
Ancestral state reconstructions
According to our BayesTraits analysis, transitions between hosts have been commonplace throughout the evolutionary history of Onchocerca (Fig. 1). This analysis indicates that there have been multiple transitions between cervid and bovid hosts. For example, the common ancestor to the extant Onchocerca was most likely associated with a cervid host (Fig. 1). Our analysis also indicates that several lineages of Onchocerca from these cervids and bovids have gone on to colonize canids, camelids, suids, and humans. For example, O. jakutensis (cervid parasite), O. fasciata (camelid parasite), and the common ancestor to O. volvulus (human parasite) and O. ochengi all appear to have evolved from different bovid-parasitizing lineages.
Our maximum likelihood ancestral state reconstructions support some, but not all of the ancestral character states from our BayesTraits analysis (Figs S1–S3 and Table S3, online version only). All of the single gene phylogenetic analyses suggested that the most recent common ancestor to O. volvulus was associated with cattle (Figs S1–S3, online version only), while the 3 gene ancestral state reconstruction suggested that cattle and humans were about equally likely to have been the ancestral host. The 12S and 16S analyses (Figs S1 and S2, online version only) suggested that O. cervipedis descended from a worm associated with cervids, but ND5 supported a bovid-associated ancestral state (Fig. S3, online version only). The 12S and ND5 analyses indicated that the most recent common ancestor to O. lienalis and Onchocerca from New York deer was associated with cattle (Figs S1 and S3, online version only), while the 16S analysis supported cervid association.
DISCUSSION
Previous studies of Onchocerca have identified multiple species associated with cervids in Eurasia, but only 1, O. cervipedis, from North America. In the present study, a relatively small number of Onchocerca obtained from white-tailed deer in New York State and moose from the Northwest Territories were examined. Our molecular phylogenetic results clearly show that there are at least 2 species of Onchocerca infecting North American cervids. The morphological match of the moose-associated species with the description of O. cervipedis suggests that the samples from moose are O. cervipedis and that worms obtained from New York deer represent a previously undescribed species of Onchocerca. Alternatively, the new species may represent a range expansion of a Eurasian Onchocerca species, or a new geographical record of a Holarctic species. Our phylogenetic analyses, however, allow us to exclude all Eurasian species except for O. garmsi, for which no sequence data exist.
The finding of 2 molecularly very distinct species of Onchocerca in a relatively small sampling scheme (moose from Northwest Territories and white-tailed deer in New York) suggests that the number of cervid-associated Onchocerca species in North America may actually be considerably higher. A greater diversity could be found for at least 3 reasons. First, different cervid species in North America may be infected with different species of Onchocerca. For instance, what has been called O. cervipedis has been reported from all species of North American cervids, including Odocoileus spp. (mule and white-tailed deer), Alces alces (moose), Cervus canadensis (elk), and Rangifer tarandus (caribou), as well as from Antilocapra americana (pronghorn), which belongs to the Antilocapridae (Hibler, Reference Hibler1965; Low, Reference Low1976; Samuel et al. Reference Samuel, Barrett and Lynch1976). However, it is possible that these worms comprise a complex of species associated with different hosts.
Second, individual host species might be associated with different species of Onchocerca in different parts of their geographical ranges, perhaps due to the occurrence of different blackfly vectors in different areas. For instance, while many records of O. cervipedis come from western North America (DeNio and West, Reference DeNio and West1942; Herman and Bischoff, Reference Herman and Bischoff1946; Hibler, Reference Hibler1965; Weinmann et al. Reference Weinmann, Anderson, Longhurst and Connolly1973), there are also reports of O. cervipedis from eastern North America (Robbins and Clark, Reference Robbins and Clark1978 and references therein). However, identifications based on microfilariae, such as those presented by Robbins and Clark (Reference Robbins and Clark1978), are difficult, and may be particularly prone to missing cryptic species.
Finally, different species of Onchocerca may be distributed differently within the bodies of their hosts. For example, red deer in Europe harbor 4 different Onchocerca species that are found in different locations within the host (Morandi et al. Reference Morandi, Krueger, Panarese, Sarli, Verin, Nicoloso, Benazzi and Galuppi2011). The O. cervipedis from moose in our study occurred in subcutaneous tissues surrounding the metatarsus, in agreement with Wehr and Dikman's (Reference Wehr and Dikmans1935) original description. However, other studies have reported O. cervipedis from other parts of the body, such as the ear from Arizona deer (Hibler, Reference Hibler1965), subcutaneous tissue from the ventral abdomen, flanks, brisket, and shoulder of Columbian black-tailed deer (Odocoileus hemionus columbianus) in California (Weinmann et al. Reference Weinmann, Anderson, Longhurst and Connolly1973), and connective tissue from the shoulder and rump of Montana deer (DeNio and West, Reference DeNio and West1942). By analogy with the situation in red deer, what has been called O. cervipedis may comprise multiple species, each associated with different parts of the host body. Therefore, more extensive molecular phylogenetic studies of Onchocerca from different host species, geographical regions, and body sites within hosts may reveal considerably greater diversity in North America than described to date.
Although we have considered only 2 cervid-associated species of Onchocerca in North America, our molecular phylogenetic analysis indicates that these 2 species are quite distantly related. Interestingly, the deer-associated species appears to be the sister group of the cattle-infecting species O. lienalis, which together belong to a clade that also includes O. ochengi and O. volvulus, the causative agent of River Blindness in humans. We found evidence of co-infection in 2 deer; however, all of the deer-associated Onchocerca sequences formed a monophyletic clade in our three-gene and ND5 phylogenies, suggesting that they belong to the same species.
Our population genetic analyses also suggest that the deer-associated Onchocerca are from a single species. The low nucleotide diversity and high haplotype diversity of the deer-associated Onchocerca is similar to that found in O. volvulus (Morales-Hojas et al. Reference Morales-Hojas, Cheke and Post2007). This pattern is suggestive of a recent expansion after a population bottleneck (Grant and Bowen, Reference Grant and Bowen1998), but the small sample size and non-significant Tajima's D results (where significant, negative values would also suggest population expansion after a bottleneck (Tajima, Reference Tajima1989)) means that this result should be interpreted with caution. Our ancestral state reconstruction suggests that the association with deer is the result of a host switch from bovids, which would be consistent with a population bottleneck and subsequent expansion. Further population genetic analyses are necessary to explore this hypothesis.
The molecular phylogenetic analyses and ancestral state reconstructions presented here agree with previous work suggesting that host-switching instead of co-speciation has influenced the evolutionary history of Onchocerca (Krueger et al. Reference Krueger, Fischer and Morales-Hojas2007; Morales-Hojas, Reference Morales-Hojas2009; Morales-Hojas et al. Reference Morales-Hojas, Cheke and Post2006). Our analyses also indicate that association with cervids and bovids is widely and deeply distributed across the phylogeny of Onchocerca. The three-gene ancestral state reconstruction is in general agreement with the single-gene ancestral state reconstructions; however it should be noted that it is likely that incomplete sampling of the genus affects these analyses. For example, the 16S-based reconstruction, which has the best coverage of suid-associated Onchocerca, suggests that the common ancestor to extant Onchocerca was associated with suids, while our other reconstructions suggests that the common ancestor associated with cervids. The results of these reconstructions may change as we gather more data and greater knowledge of Onchocerca diversity.
The Onchocerca phylogenetic hypotheses presented here are in general agreement with previous molecular phylogenetic studies of Onchocerca (Krueger et al. Reference Krueger, Fischer and Morales-Hojas2007; Morales-Hojas, Reference Morales-Hojas2009; Morales-Hojas et al. Reference Morales-Hojas, Cheke and Post2006). Our analyses showed strong support for a monophyletic clade containing the African species O. volvulus, O. ochengi, O. dukei, and 2 unidentified species. The nodes within this clade, however, were weakly supported, as were most of the deeper nodes within our trees. Our three-gene analysis placed O. ramachandrini (parasite of African suid, see Wahl, Reference Wahl1996) as sister to the rest of the genus. Our single gene trees placed either O. dewittei (parasite of Asian suid, see Uni et al. Reference Uni, Bain, Takaoka, Miyashita and Suzuki2001) or O. ramachandrini as sister to the rest of the genus. All of these placements, however, were poorly supported. Although morphology and diversity both suggest that Onchocerca arose in Africa (Morales-Hojas, Reference Morales-Hojas2009), the molecular phylogenetics presented in our study and previous studies have been unable to either verify or reject this suggestion.
Our phylogenetic analysis included an estimation of host-association probabilities at various nodes in the tree, thus enabling the inference of host shifts. For example, both our Bayesian and likelihood ancestral state reconstructions suggest that the human parasite O. volvulus represents a host switch from bovids, although the Bayesian analysis does not distinguish between the switch occurring directly or in the common ancestor to O. volvulus and O. ochengi. Our analyses also suggest that transitions between cervid and bovid hosts are particularly common, and that cervids and bovids appear to act as springboards for colonization of other groups such as camels, humans, suids, and canids. In general, our ancestral host analysis indicates that cervids and bovids have probably been key hosts throughout most of the evolutionary history of Onchocerca.
Although rare, reports of zoonotic onchocerciasis are on the rise (Uni et al. Reference Uni, Boda, Daisaku, Ikura, Maruyama, Hasegawa, Fukuda, Takaoka and Bain2010; Fukuda et al. Reference Fukuda, Otsuka, Uni, Boda, Daisaku, Hasegawa, Takaoka and Bain2011; Takaoka et al. Reference Takaoka, Fukuda, Otsuka, Aoki, Uni and Bain2012). If the deer-associated Onchocerca in New York are vectored by a biting insect that feeds on both deer and humans, such as Simulium species, several factors indicate that zoonotic infections may be possible. The deer population in New York state increased dramatically within the last century, and is estimated to have peaked at over one million in 2000–2002, after which it has declined somewhat (Bifaro et al. Reference Bifaro, Clark, Clarke, Dente, DiDonato, Farquhar, Heerkens, Hurst, Kautz, Kirsch, Peil, Reed, Rielhman, Spierto and Swift2011). The overall prevalence of Onchocerca infection among our white-tailed deer samples, taken from widely separated regions within New York, was 23%. If these samples are representative of the population as a whole, then close to a quarter-million deer in New York are currently infected with Onchocerca. The widespread distribution and high population densities of white-tailed deer, in conjunction with the relatively high prevalence of Onchocerca infection in New York deer, suggest that deer could be an important ecological source of zoonotic infections or potential colonization of other host species, including humans. The occurrence of what are likely to be obligate symbiotic Wolbachia within deer-associated Onchocerca suggests that antibiotic treatment might be effective, should zoonotic infections of humans occur (Pfaar and Hoerauf, Reference Pfaar and Hoerauf2006).
In conclusion, we have shown that the Onchocerca parasites of cervids in North America include at least 2 distantly related species, contrary to previous reports of only a single species. Molecular phylogenetic studies integrated with classical parasitological techniques and targeting more host species across a greater geographical area will be required to estimate the full diversity of Onchocerca species infecting cervids and other wild ungulates in North America. Our ancestral state reconstructions showed that host association is evolutionary labile in Onchocerca and that cervid- and bovid-associated species have likely been an important source for shifts to other host groups. Thus, a better understanding of cervid-associated Onchocerca in North America could yield insights into possible host shifts to other species in the future.
ACKNOWLEDGEMENTS
We thank Dick Simonson and Glens Falls Animal Hospital for their generous assistance in acquiring deer samples. We are especially grateful to Mr Simonson, the project would not have been possible without his help. We also thank Ed Cupp, Danny Mead, and Ben Makepeace for sharing their protocols and advice, Susan Kutz (Faculty of Veterinary Medicine, University of Calgary) for support in obtaining the moose-derived specimens, and two reviewers for helpful comments on the manuscript. This research was supported by GCE grant OPP1017537 from the Bill & Melinda Gates Foundation.






