


INTRODUCTION
Whirling disease and its associated myxosporean agent, Myxobolus cerebralis, were first described in Europe in 1898 among farmed rainbow trout (Hofer, 1903). The disease spread throughout Europe and eventually to the USA through the international fish trade (Hoffman, 1970; El-Matbouli, Fischer-Scherl and Hoffmann, 1992). Whirling disease is considered not only a problem in the fish culture industry but is also a major threat to the survival of wild rainbow trout in North America (Hedrick et al. 1998). The disease has been recognized as a central cause of the catastrophic decline of wild rainbow trout populations in the states of Idaho, Montana, Colorado and Utah, USA (Nehring and Walker, 1996; Hedrick et al. 1998). Brown trout are considered the natural host of M. cerebralis, for even though they become infected, they remain asymptomatic (Hoffman, Dunbar and Bradford, 1962). The severe decline in wild rainbow trout populations has stimulated a renewed interest in exploring the pathobiology and host-parasite interaction of M. cerebralis (El-Matbouli et al. 1999).
Myxobolus cerebralis requires an invertebrate host to complete its life-cycle and only a single species of oligochaete worm is susceptible, Tubifex tubifex (Markiw and Wolf, 1983). The parasite alternates between 2 spore forms: an actinosporean (triactinomyxon) that develops in the oligochaete, and a myxosporean (Myxobolus) in salmonid fish (Wolf and Markiw, 1984; El-Matbouli and Hoffmann, 1998; El-Matbouli, Hoffmann and Mandok, 1995). M. cerebralis attacks young rainbow trout before their cartilage hardens to bone, causing skeletal deformities and neurological disruption giving rise to the disease's chief symptoms of black tail, tail-chasing behaviour (whirling) and cranial, jaw and opercular deformities (Schäperclaus, 1931).
From a molecular biological point of view, little is known about the genetics of either triactinomyxon or Myxobolus spore stages. The sequence of the parasite's small subunit 18S ribosomal DNA gene (18S rDNA) is known, as are some other sequences coding for actins and proteases: M. cerebralis beta-actin gene (Kelley, Beauchamp and Hedrick, 2004a); 18S ribosomal RNA gene; internal transcribed spacer 1, 5·8S ribosomal RNA gene, and internal transcribed spacer 2 (Whipps et al. 2004a); 28S ribosomal RNA gene (Whipps et al. 2004b); cathepsin Z-like cysteine proteinase gene (Kelley et al. 2003) and chymotrypsin-like serine protease (Kelley et al. 2004b).
Without the parasite's complete DNA sequence or some other point of reference, we can't know if a discovered gene even belongs to M. cerebralis. This paucity of information is an obstacle to researchers seeking to probe specific M. cerebralis genes and their functions.
Construction of a cDNA library for the parasite would therefore represent a major research advance, and is the central goal of this study. The library will establish a genetic information base for M. cerebralis, containing protein-encoding sequences from the genome, which can then be used by researchers to analyse functions of specific genes.
MATERIALS AND METHODS
Experimental infection of oligochaetes and collection of triactinomyxon spores
Myxobolus cerebralis myxospores were obtained from the skulls of clinically diseased rainbow trout, according to the method of El-Matbouli et al. (1995), and counted. Approximately 10000 (150 g) infection-free T. tubifex, from SPF culture, were kept in a 10 litre plastic aquarium, in a 5 cm layer of sterilized sand covered with aerated, de-chlorinated tap water at 14–15 °C, and fed Algamac-2000. The T. tubifex were exposed to approximately 4×106 viable M. cerebralis, a dose of about 400 spores/worm (El-Matbouli et al. 1999). The water from the culture was examined weekly for the presence of triactinomyxon spores. Following detection of waterborne triactinomyxon spores approximately 3 months post-exposure, the water over the culture was siphoned off purified with Percoll gradient and counted. The purified triactinomyxon spores were then concentrated and re-suspended in a small volume of distilled water and kept at −20 °C until required or kept in RNAlater for isolation of RNA.
Selection of highly infected Tubifex tubifex
When the infected T. tubifex reached their period of peak release of triactinomyxon spores between 140 and 160 days, individual worms were separated into multi-well plates with de-chlorinated water and maintained for approximately 1 week at 14–15 °C. This was both to determine which worms were releasing spores, and which were most productive. Worms that release high numbers of spores were selected and preserved in RNAlater for using in RNA extraction.
Extraction of total RNA
Total RNA, from (70) oligochaetes producing the highest numbers of triactinomyxons, was isolated following the RNeasy Mini protocol for isolation of total RNA from animal tissues. Thirty mg of RNAlater-stabilized samples were placed in liquid nitrogen, and thoroughly ground with a mortar and pestle. The tissue powder and liquid nitrogen was decanted into a liquid-nitrogen-cooled 2 ml tube, and the nitrogen allowed to evaporate. Samples were then resuspended in an appropriate volume of guanidine isothiocyanate buffer, and homogenized by passing the lysate 5 times through a 20-gauge needle. Then 70% ethanol was added to the lysate to create conditions that promote selective binding of RNA to the RNeasy silica-gel membrane. The sample was then applied to the RNeasy mini–column, the RNA allowed to bind before being rinsed and then eluted in RNase-free water. The concentration of RNA was determined by measuring the absorbance at 260 nm (A260), while the purity of the RNA was determined by measuring the absorbance at 260 nm and 280 nm (A260/A280) in a spectrophotometer.
Isolation of messenger RNA (mRNA) from total RNA
After extraction of RNA from the oligochaetes, mRNA was isolated from the total RNA using the Oligotex mRNA spin-column protocol as per manufacture's instructions. The concentration of the mRNA was measured with a spectrophotometer. Prime RNase inhibitor was added to the mRNA to protect it from RNase contamination.
Construction of the cDNA library
The cDNA library was constructed with the ZAP Express™ cDNA synthesis kit with some modification: 5 μg Poly A+ RNA was primed in the first strand cDNA synthesis with the oligo (dT) linker-primer (Table 1) that contains an XhoI restriction site, then transcribed using StrataScript™ reverse transcriptase (50 U/μl) at 42 °C for 1 h. The second strand cDNA was generated using RNase H and DNA polymerase I. After blunting of the cDNA termini by cloned Pfu DNA polymerase; the reaction was purified and concentrated using MinElute™ gel extraction kit protocol as per the manufacture's instructions. cDNA was then lyophilized and resuspended in EcoRI adapters which ligated overnight at 8 °C by using T4 DNA ligase.
Table 1. Sequences of primers used (N.B. N=A, C, G or T; N−1=A, G, or C; V=A, C, G.)

The cDNA was then incubated with T4 polynucleotide kinase at 37 °C for 30 min to phosphorylate the EcoRI ends; and then digested with XhoI enzyme. The reaction then was cleaned up and concentrated using MinElute™ gel extraction kit. The cDNA was lyophilized and resuspended in 50 μl 1× STE buffer (150 mM NaCl, 10 mM Tris-HCl, pH 7·4, 1 mM EDTA) and subjected to size fractionation using SizeSep™ 400 Spin Column, Sepharose CL-4B. The collected fraction was concentrated by MinElute™ gel extraction kit, lyophilized and then resuspended in 5 μl of sterile water. After ligation of the constructed cDNA into ZAP Express vector by T4 DNA ligase enzyme at 4 °C for 2 days, the resulting DNA was packaged in vitro using Gigapack III gold packaging extract at room temperature for 2 h. Then 20 μl of chloroform and 500 μl of SM buffer (NaCl, Tris-HCl, pH 7·5, MgSO4 Gelatine) were added to the packaging extract and mixed gently, and the tube centrifuged briefly. The supernatant containing the phage was kept at 4 °C until titred.
Titration of the primary cDNA library
A culture of XL1-Blue MRF

host strain, in LB medium, supplemented with 10 mM MgSO4 and 0·2% maltose, was grown to an OD600 of 1·0, then the bacteria were pelletized by centrifuging at 1000 g for 10 min. The cells were gently resuspended to half the original volume with sterile 10 mM MgSO4, and diluted to a final OD600 of 0·5. LB top agar was melted and cooled to 55 °C in a water bath. Then 1 μl of lambda phage packaging material was added to 300 μl of the diluted host cells. Also, 1 μl of 1[ratio ]10 dilution of packaging material in SM buffer was added to 300 μl of host cells. The phage and bacteria were incubated for 15 min at 37 °C to allow the phage to attach to cells. LB top agar, 0·5 M isopropyl-beta-D-thiogalactopyranoside (IPTG) (in water) and X-gal (250 mg/ml in dimethylformamide (DMF)) were then added and the solution was mixed, and poured immediately onto LB agar plates, pre-warmed to 37 °C, and distributed evenly across the surface of the plate, before being left to solidify at room temperature. The plates were incubated at 37 °C overnight to develop the plaque colour: background plaques are blue, while recombinant plaques are white. After incubation, the plaques were counted and the ratio of blue to white was calculated to determine the library titre.
Antibody screening of the cDNA library
Immunoscreening of the constructed ZAP Express cDNA library was performed by use of the anti-triactinomyxon polyclonal antibody and ECL™ chemiluminescent detection method. Briefly, E. coli XL1-Blue MRF
were infected with 1000 phages containing cDNAs, plated onto pre-warmed LB–agar plates and incubated for ~3·5 h at 42 °C. After about 3·5 h plaques would begin to form, and the plates were overlaid with the 10 mM IPTG-soaked nitrocellulose filters and incubated at 37 °C overnight. Filters were removed carefully and washed 3×10 min, with (TBST) Tris-buffer saline with Tween 20 (150 mM NaCl, 10 mM Tris-HCl, pH 8·0, 0·05% Tween 20). They were then blocked with 5% skimmed milk powder in TBST at 4 °C for 2 h with agitation, and incubated overnight with the anti-triactinomyxon polyclonal antiserum (pre-absorbed against E. coli proteins and diluted 1[ratio ]1000 in TBST). Filters were washed 3 times in TBST and reacted with alkaline phosphate-conjugated anti-rabbit IgG (diluted in TBST according to the manufacture's instructions) for 5 h at 4 °C. Membranes were given a final wash in TBST and were developed with ECL™ chemiluminescence as per the manufacture's instructions.
Plaques identified as positive by antibody screening were subjected to secondary and tertiary immunoscreening. Individual plaques were selected randomly after tertiary screening and used as a template for PCR for amplification of cDNA insert fragments using the T3 and T7 primers. Each PCR was performed in a 50 μl final volume containing 20 μM of each primer, 47 μl of Reddy Mix PCR Master Mix (75 mM Tris-HCl, pH 8·8, 1·25 U Taq Polymerase, 20 mM (NH4)2SO4, 1·5 mM MgCl2, 0·01% Tween 20, 0·2 mM each dNTPs) and seeded with 1 μl of phage. PCR parameters were as follows: an initial denaturation at 95 °C for 3 min, followed by 35 cycles of 30 s at 95 °C, 45 s at 48 °C and 30 s at 72 °C. Final extension was carried out at 72 °C for 10 min. Amplified products were visualized on 1% agarose gels and compared against DNA molecular weight standard.
pBK-CMV phagemid, in vivo excision, plasmid DNA isolation and cDNA insert size estimation
After cDNA clones of interest were identified in the intact Lambda ZAP Express vector by antibodies, a representative clone was selected at random (45-5) to test the specificity of the library to triactinomyxon spores. The selected plaque was purified to homogeneity and corresponding pBK-CMV phagemid was rescued by in vivo excision using ExAssist helper phage. The E. coli XLOLR cells were infected with the phagemid and were plated onto LB-kanamycin (50 mg/ml) agar plates and incubated at 37 °C overnight. Colonies appearing on the plate containing the pBK-CMV double-stranded phagemid having the cloned DNA insert. Plasmid DNA was isolated from the grown colony, after its overnight propagation in LB/kanamycin (50 μg/ml) broth, by using QIAprep spin miniprep kit (Qiagen GmbH, Hilden, Germany) according to the manufacture's instructions. The plasmid DNA was used as a template for PCR reaction to estimate the size of the cDNA fragments insert using the T3 and T7 primers (Table 1) as previously described.
DNA sequencing, sequence analysis and primer construction
The purified plasmid DNA was sequenced (SEQLAB, Göttingen, Germany). A National Centre for Biotechnology Information (NCBI) non-redundant nucleotide BLASTn search was carried out to find any similarities to the edited sequences (Altschul et al. 1990). After sequencing and alignment of the clone, primers 46-5 for and 46-5 rev were designed using ‘Oligo’ software to ensure that their sequence belonged to M. cerebralis and not to the worm. Three other primers, GSP1, GSP2 (both 5′ end) and GSP5 (3′ end) were designed using the same software, for use with RACE amplification of the ends of the cDNA, to obtain the sequence of the entire clone. The primer sequences are illustrated in (Table 1).
5′ and 3′ RACE amplification
The first-strand cDNA was primed with the constructed GSP1 primer and 20 U AMV reverse transcriptase at 55 °C for 60 min. After purification of the PCR product by the high pure PCR product purification kit, a known sequence was added to the purified 3′ end by incubation with terminal transferase enzyme at 37 °C for 30 min in the presence of 2 mM dATP and 10×reaction buffers. The tailed cDNA was amplified by PCR using the GSP2 and dT-anchor primers in a 50 μl reaction volume comprising: 7 μl of dA-tailed cDNA, 37·5 μM oligo dT-anchor primer, 12·5 μM GSP2 primer, 1 μl of deoxynucleotide mixture, 2·5 U AmpliTaq® DNA polymerase, 5 μl of 10× reaction buffer and PCR-grade water to 50 μl. The PCR used the following cycle conditions: 94 °C for 2 min, 35 cycles of 94 °C for 15 s, 63 °C for 30 s and 72 °C for 1 min, with final elongation of 72 °C for 7 min. First strand synthesis for the 3′ RACE was primed with oligo dT-anchor primer and reverse transcriptase enzyme, and then the cDNA was amplified by using the GSP5 and PCR anchor primers. Then 20 μl of each 3′ and 5′ PCR product was analysed on a 1% ethidium bromide-stained agarose gel with a corresponding DNA molecular weight ladder. The corresponding (single) bands excised and purified using MinElute gel extraction kit (Qiagen) as per the manufacture's instructions.
Cloning and sequencing of the RACE products
To facilitate sequencing, purified 5′ and 3′ RACE products were cloned into the pCR® 4-TOPO® vector using TOPO TA cloning® kit (Invitrogen, Karlsruhe, Germany), according to the manufacture's instructions. Recombinant plasmids were purified from Escherichia coli using QIAprep Spin Miniprep kit, as per manufacture's instructions. Plasmid concentrations were determined by spectrophotometer. The cloned RACE products insert was sequenced (SEQLAB, Göttingen, Germany). BLASTn and BLASTx searches were conducted, respectively, to confirm whether the sequences were unique and to search for translation of the sequence and protein similarities (Altschul et al. 1990).
RESULTS
Construction of triactinomyxon spore cDNA library
A triactinomyxon spore cDNA library was successfully constructed. The titre of the primary library was 0·5×106 and 97% of the library (4·85×105) produced recombinant white plaques.
Immunological screening of the constructed cDNA library
Primary screening of the library revealed only a few positive plaques out of the 1000 plaques on the plate (Fig. 1). Significantly higher yields were obtained in the secondary screening, following propagation of positive clones (Fig. 2). For tertiary screening, a phage plug was removed and diluted into SM buffer before being plated at a low titre of 20–50 particles/plate that allowed single plaques to be isolated (Fig. 3). Approximately 526 positive plaques from triactinomyxon spores were detected by immunological screening with anti-triactinomyxon antibodies. The PCR result of a subset of these positive plaques revealed that positive clones contained inserts with different molecular weights, ranging from 650 bp to 2 kb, indicating successful construction of the library.
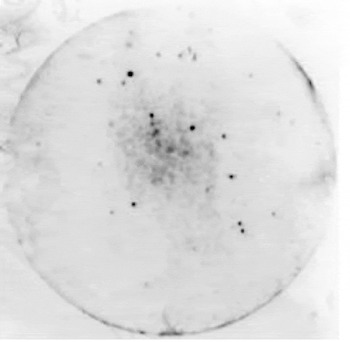
Fig. 1. ECL-Hyperfilm showing primary screening of the cDNA library with positive plaques visible as black spots on the plate.
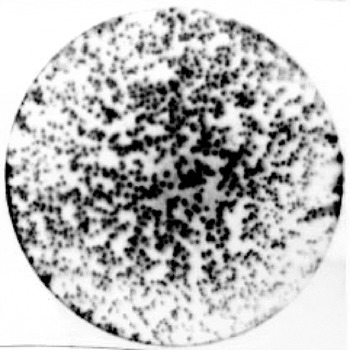
Fig. 2. ECL-Hyperfilm showing the result of secondary screening of the positive plaques of the cDNA library, positive plaques visible as black spots on the plate.

Fig. 3. ECL-Hyperfilm showing the result of tertiary screening of the cDNA library positive plaques, positive plaques visible as black spots on the plate.
Size estimation and specificity of the selected clone
The PCR amplified a 750 bp segment representing the cDNA insert of the randomly selected clone 46-5. The specificity of this clone to triactinomyxon spores was confirmed by amplification of the 511 bp fragment of cDNA from triactinomyxon spores using the 46-5 specific primer pair; while there was no amplification of non-infected oligochaetes cDNA or the negative control.
Sequence analysis of the full-length cDNA clone 46-5. After performing the RACE reaction, the complete sequence of the clone 46-5 is 1461 bp long (Fig. 4). The sequence was deposited in GenBank with an Accession number DQ138322. A nucleotide-nucleotide BLASTn and protein-protein BLASTp search demonstrated a high percentage similarity of the nucleotide and amino acid sequence respectively with ribosomal protein L23a and 60S ribosomal protein L23a. A coding sequence (800–1375 bp) was detected by the GENSCAN program (Fig. 4). The predicted peptide sequence of the detected coding sequence (Fig. 5) revealed, after alignment, the same identity with the ribosomal protein L23a, as with the whole amino acid sequence alignment (Fig. 6). Also a conserved domain of ribosomal protein L23 was detected after alignment of the predicted peptide sequence with the protein family data base (Fig. 7).

Fig. 4. The complete nucleotide sequence of full-length cDNA clone 46-5 (GenBank Accession number DQ138322). Numbers on the right denote base positions. The underlined sequences represent the coding sequences detected by the GENSCAN software (800–1375).

Fig. 5. Peptide sequence of the coding sequence 800–1375 bp of the clone 46-5. Numbers on the right denote peptide positions.

Fig. 6. Alignment of the amino acid sequences of the clone 46-5 with GenBank database. Numbers on the right and left are the location of amino acids on the sequence. Arg. irrad=Argopecten irradians (AAN05592), Branch. Belch=Branchiostoma belcheri (AAN52376), Ictalu. Punc=Ictalurus punctatus (AAK95150) and Tetra. Nigro=Tetraodon nigroviridis (CAG00513).

Fig. 7. Alignment of the amino acid sequences of the coding sequences of the clone 46-5 with the protein family (pfam) data base which detected the conserved domain of ribosomal protein L23a. Numbers on the right and left are the location of amino acids on the sequence.(-) indicate a gap. klu lac=Kluyveromyces lactis (P48045), Meth jan=Methanocaldococcus jannaschii (P54016), Halob salin=Halobacterium salinarum (Q06842), Mycop capri=Mycoplasma capricolum (P10140), Geob stea=Geobacillus stearothermophilus (P04454), Odon sin=Odontella sinensis (P49559), Mycop genit=Mycoplasma genitalium (P47399) and Eug lon=Euglena longa (P34771).
DISCUSSION
This study involved construction of a cDNA library from triactinomyxon spores, which can be searched for genes expressed in the triactinomyxon spore stage. Total RNA was extracted from the highly triactinomyxon spores producing worms (about 120 days post-exposure to M. cerebralis spores) to increase the chance of obtaining cDNA clones from nearly all developmental stages of the parasite in the oligochaetes.
An essential step when preparing the mRNA was to separate it from the transfer RNA (tRNA) and ribosomal RNA (rRNA) fractions of total RNA. The amount of rRNA and tRNA is vastly greater than mRNA and will decrease the efficiency of the reverse transcription reaction; which is also why cDNA is constructed from mRNA not from total RNA. Fortunately, the bulk of mRNA carries strings of poly-As at their 3′ termini, and can be separated from total RNA on an oligo (dT) cellulose column (Aviv and Leder, 1972; Chomczyniski and Sacchi, 1987). The oligo (dT) linker primer used in the first strand synthesis 5′-GAGAGAGAGAGAGAGAGAGAACTAGTCTCGAGTTTTTTTTTTTTTTTTTT-3′ was designed with a ‘GAGA’ sequence to protect the XhoI restriction enzyme recognition site ‘CTCGAG’ and 18-base poly (dT) sequence. The restriction site allows the finished cDNA to be inserted into the ZAP Express vector in a sense orientation (EcoRI-XhoI) with respect to the lac Z promoter (Short et al. 1988), while the 18-base poly (dT) region binds to the 3′ poly (A) region of the mRNA template. The Strata™ Script enzyme is a genetically engineered Moloney Murine leukaemia virus reverse transcriptase without any detectable RNase H activity. The total yield of first-strand cDNA is substantially higher with this enzyme than with non-engineered reverse transcriptase and the proportion of full-length cDNAs is significantly greater (Kotewicz et al. 1988; Gerard and D'Alessio, 1993; Telesnitsky and Goff, 1993).
To protect cDNA from digestion, 5-methyl dCTP was used during the first-strand synthesis; i.e. the nucleotide mixture for the first-strand synthesis contained normal dATP, dGTP, dATP plus the analogue 5-methyl dCTP. The completed first-strands then have a methyl group on each cytosine base which protects the cDNA from restriction enzymes used in subsequent cloning steps (Han and Rutter, 1988; Huse and Hansen, 1988). Only the unmethylated site within the linker-primer was cleaved by XhoI.
The product of the first-strand synthesis, a cDNA-mRNA hybrid, was used for second-strand synthesis. RNase H produced nicks and gaps in the mRNA strand of the hybrid, creating a multitude of fragments which serve as primers that are used by DNA polymerase I during the synthesis of the second cDNA strand (Okayama and Berg, 1982; Gubler and Hoffman, 1983). The second-strand nucleotide mixture was supplemented with additional dCTP to reduce the probability of residual 5-methyl dCTP becoming incorporated in the second strand, ensuring that the restriction sites in the linker-primer will be susceptible to future restriction enzyme digestion. Uneven termini of the double-stranded cDNA were ‘nibbled’ or filled in with cloned Pfu DNA polymerase to create a blunted ends for EcoRI adapters' ligation. The adapters composed of 10-mer 3′-GCCGTGCTCCp-5′ and 14-mer oligonucleotides 5′-OH-AATTCGGCACGAGG-3′ which are complementary to each other with an EcoRI cohesive end (AATTC). The 10-mer oligonucleotide was phosphorylated (p) which allowed it to ligate to blunt termini of cDNA and other adapters; while the 14-mer oligonucleotides was kept dephosphorylated (OH) to prevent ligation to other cohesive ends.
After ligation of the adapters and inactivation of the ligase enzyme, the 14-mer oligonucleotide was phosphorylated to enable its ligation to dephosphorylated vector arms. The ligation reaction was carried out in a small volume to maintain a high concentration of adapters in order to minimize blunt-end ligation of the cDNA (Sambrook and Russell, 2001). XhoI digestion released the EcoRI adapter and residual linker-primer from the 3′ end of the cDNA and thereby prepared it for insertion into the vector. This strategy greatly improved the efficiency of the ligation step in cDNA cloning and eliminated the need to digest the cDNA with restriction enzyme before insertion into the vector (Yang et al. 1986; Elledge et al. 1991). Before insertion, the cDNA was fractionated by gel filtration to remove unused adapters and residual linker-primer created by the XhoI enzyme digestion. This process significantly increases the number of recombinants that contain cDNA (Sambrook and Russell, 2001). Fractionation also enabled fragments less than 400 bp to be discarded; these were unwanted products of incomplete first- and/or second-strand synthesis. Discarding small fragments also reduces the number of recombinants that must be screened, and increases the chance of isolating full-length cDNAs corresponding to rare mRNAs encoding large proteins (Sambrook and Russell, 2001).
The size-fractionated cDNA was next ligated to the ZAP Express vector which increases both cloning capacity and the number of unique lambda cloning sites (Jerpseth et al. 1992). Bacteriophage lambda was used as the ZAP Express vector as it is highly efficient at becoming infected by phage particles packaged in vitro. Inserts cloned into the vector can be rapidly excised out of the phage in the form of the kanamycin-resistant pBK-CMV phagemid vector allowing insert characterization in a plasmid system (Short and Sorge, 1992; Alting-Mees et al. 1992; Short et al. 1988). In addition, clones in the vector can be screened with either DNA probes or antibody probes.
The lambda library was then packaged in vitro by adding the recombinant lambda DNA to an E. coli extract containing assembly proteins and precursors required for encapsulating lambda DNA to produce infectious recombinant lambda phage (Becker, Marko and Gold, 1977). The highly efficient Gigapack III Gold packaging system was used (Kretz et al. 1994). The library was plated using an E. coli host strain, XL1-Blue MRF
, deficient in the mcr system (modified cytosine restriction) which normally cleaves DNA at methylcytosine residues (Raleigh et al. 1988; Woodcock et al. 1989).
The primary library titre was 0·5×106 with 97% recombinant plaques; a result comparable with that of Stürzenbaum et al. (2003) who constructed an earthworm cDNA library using the same protocol, and achieved a titre of 0·5×106 recombinant plaques. Similarly, a cDNA expression library constructed for sheep louse Bovicola ovis had a primary titre of 5×105, with 97% recombinant plaques (Nattrass and James, 2001).
After construction, cDNA libraries are typically amplified to provide a near limitless source of cDNA clones. This was not done for the triactinomyxon cDNA library in order to preserve accurate representation of the mRNA in the cDNA library (Sambrook and Russell, 2001). As the library was constructed from mRNA extracted from infected worms, it contains clones of both triactinomyxon spores and the host. Even a single round of amplification could distort the representation of cDNA clones through variations in growth rate of recombinants skewing the proportions of cDNA in the library. After titrating, the library was subjected to immunological screening. The chemiluminescence detection method was used to select 526 positive plaques which contained the cloned triactinomyxon mRNA. These plaques were screened 3 times to screen out any contamination by negative plaques. In the primary screening, the antiserum selected only a few positive plaques as the proportion of triactinomyxon plaques was originally low compared with oligochaete plaques. In the second screening, after selective propagation of plaques from round one, the number of positive plaques was much higher. In tertiary screening the cultures were diluted to reduce the number of plaques on the plate to allow easy selection of only single plaques. Positive clones were tested by PCR, which showed that these clones contained inserts with different molecular weights, eliminating the probability of selecting a false positive plaque that has no insert and indicating the successful construction of the library.
After in vivo excision of the clone 46-5; the XLOLR plasmid DNA containing the insert was extracted and sequenced. Specific primers were designed to amplify only triactinomyxon cDNA and not cDNA from the oligochaete host. To obtain the full length of the 46-5 cDNA, rapid amplification was used on the mRNA template between a defined internal site, using gene-specific primers constructed from the 46-5 clone, and unknown sequences at either the 3′ or 5′ end of the mRNA (Frohmann, 1994).
Nucleotide and amino acid sequencing revealed that 46-5 coded for ribosomal protein L23a, and comparison with the database showed that sequence homology compared with different organisms ranged from 60 to 85%: 85% with Oncorhynchus mykiss, 82% with Argopecten irradians, 78% with Branchiostoma belcheri, 73% with Danio rerio, 65% with Homo sapiens, and 60% with Spincia deracea.
The GENSCAN program detected coding sequences of 575 bp (from 800 bp–1375 bp) with prediction of the peptide sequence of these coding sequences. These peptide sequences, after alignment by BLASTp search, revealed homology with the ribosomal protein L23 from different organisms as: 72% identity with Tetraodon nigroviridis, 73% with Ictalurus punctatus, 76% with Branchiostoma belcheri and 78% with Argopecten irradians. A conserved domain to ribosomal protein L23 was detected during the alignment process of the triactinomyxon (clone 46-5) predicted peptide sequence in GENBANK (Marchler-Bauer et al. 2003). This putative conserved domain was detected after alignment of the peptide sequence with the protein families' database of alignments and hidden Markov models (pfam) which produced 60% alignment with the CD of the ribosomal protein L23 family (pfam): Kluyveromyces lactis (GenBank Accession number P48045), Methanocaldococcus jannaschii (GenBank Accession number P54016), Halobacterium salinarum (GenBank Accession number Q06842), Mycoplasma capricolum (GenBank Accession number P10140), Geobacillus stearothermophilus (GenBank Accession number P04454), Odontella sinensis (GenBank Accession number P49559), Mycoplasma genitalium (GenBank Accession number P47399) and Euglena longa (GenBank Accession number P34771).
The BLASTp search using the peptide sequence revealed alignments with a high percentage similarity (60–85%) to ribosomal protein L23a and 60S ribosomal proteins of different organisms. These results strongly suggest that the triactinomyxon sequence 46-5, codes for a functional protein belonging to the same family as ribosomal protein L23.
Ribosomes are large ribonucleoprotein complexes present in all living cells, which are responsible for manufacturing proteins translated from mRNA blueprints (Maguire and Zimmermann, 2001). Ribosomes are divided into large- and small-subunits (Öhman et al. 2003); clone 46-5 coded for ribosomal protein L23 (denoted as L25 in some eukaryotes) which is a component of the large-subunit (Öhman et al. 2003). The function of L23 has been recently identified as the anchor point for the signal recognition particle, positioning the particle in such a way that it can read the signal sequence of the nascent polypeptide (Pool et al. 2002). Kramer et al. (2002) showed that L23 can have an additional function to provide a docking site for the Trigger Factor chaperone which assists the folding process of newly synthesized polypeptides. As ribosomal proteins are located near the opening of the exit tunnel it suggests a possible role in protein secretion, since such ribosomal proteins are in a position to associate with the translocon of the endoplasmic reticulum during protein secretion (Nissen et al. 2000). L23 ribosomal protein is essential for the growth of E. coli and the association of Trigger Factor with the ribosome. Mutation of an exposed glutamate in L23 prevents Trigger Factor from interacting with ribosomes and nascent chains, and leads to protein aggregation and conditional lethality in cells which lack the protein-repair function of the DnaK chaperone (Kramer et al. 2002). Eukaryotic ribosomal subunits L23a and L35, which are close to the nascent-chain exit site, have been shown to comprise the ribosome attachment site for signal recognition particle 54, SRP54 (Pool et al. 2002). SRP54 is therefore strategically positioned to scan emerging polypeptides for the presence of a signal sequence (Willem, de Gier and Luirink, 2003). In E. coli, L23 (a homologue of L23a) seems to have an even more intricate role, functioning as an attachment site not only for the SRP (Gu et al. 2003; Ullers et al. 2003) but also for the chaperone and prolyl-isomerase trigger factor (Kramer et al. 2002).
Given the universal requirement for ribosome function, it is not surprising that the ribosomes of all organisms, both eukaryotic and prokaryotic, are highly similar. Accordingly, some ribosomal constituents have structural features that are highly conserved between species and across kingdoms (McIntosh and Bonham-Smith, 2001). Further research would be required to investigate the specific function of triactinomyxon ribosomal protein L23.
This study was supported in part by the Egyptian government as a full-term mission and the Whirling Disease Foundation and U.S. Fish and Wildlife Service.








