INTRODUCTION
Fatty acid binding proteins (FABPs) are key molecules for the facilitation of fatty acid uptake and transport. In general, cytoplasmic FABPs have molecular masses of 14–15 kDa and have a highly conserved tertiary structure consisting of 10 anti-parallel β-strands and 2 short α-helices. At least 9 tissue-specific FABP types have been identified in vertebrates and several putative physiological functions of vertebrate FABPs have been proposed including the promotion of cellular uptake and intracellular trafficking of fatty acids and the involvement in modulation of mitosis, cell growth and differentiation (for reviews see Zimmerman and Veerkamp, Reference Zimmerman and Veerkamp2002; Chmurzynska, Reference Chmurzynska2006). Besides that, FABPs have been suggested to participate and/or modulate fatty acid-mediated signal transduction pathways, and fatty acid regulation of gene expression, and to prevent local high fatty acid concentrations that cause a toxic effect to the cell (Haunerland and Spener, Reference Haunerland and Spener2004). However, the understanding of the definite function of each FABP type and also their potential mechanism of action has just begun to arise in recent years (for recent reviews see Storch and Corsico, Reference Storch and Corsico2008; Storch and McDermott, Reference Storch and McDermott2009).
Investigation of invertebrate FABPs, while not as extensive as in vertebrates has also been steady, with an increasing number of proteins isolated and characterized. A review that summarizes the available data and elucidates the relationship of FABPs with their vertebrate counterparts has been recently published by Esteves and Ehrlich (Reference Esteves and Ehrlich2006). In trematodes FABPs are essential as the parasites are incapable of synthesizing the full set of necessary lipids de novo (Meyer et al. Reference Meyer, Meyer and Bueding1970). The first FABP cDNAs were isolated from S. mansoni (Sm14) and F. hepatica (Fh15) in 1991 and 1992 (Moser et al. Reference Moser, Tendler, Griffiths and Klinkert1991; Rodriguez-Perez et al. Reference Rodriguez-Perez, Rodriguez-Medina, Garcia-Blanco and Hillyer1992) a few years after the native Fasciola proteins were recognized as cross-protective antigens in these genera (Hillyer, Reference Hillyer1985; Hillyer and Soler de Galanes, Reference Hillyer and Soler de Galanes1988). The protective properties were later confirmed in vaccine studies with the schistosome FABPs (Tendler et al. Reference Tendler, Vilar, Brito, Freire, Katz and Simpson1995, Reference Tendler, Brito, Vilar, Serra-Freire, Diogo, Almeida, Delbem, Da Silva, Savino, Garratt, Katz and Simpson1996). It was then established that the purified native FABP complex, denoted Fh12 was not identical to the protein encoded by the cloned cDNA (Hillyer, Reference Hillyer1995) and follow-up analyses of Fh12 showed that it contained at least 8 FABP isoforms which differed in their isoelectric points and also caused different immune responses in rabbits (Espino and Hillyer, Reference Espino and Hillyer2001; Espino et al. Reference Espino, Rodriguez Medina and Hillyer2001). Several later studies demonstrated the efficacy of FABPs as a vaccine, and the schistosome FABP Sm14 has recently been selected for clinical trials (Tendler and Simpson, Reference Tendler and Simpson2008).
It is, therefore, of interest to further investigate the roles of the various FABP isoforms to better understand their impact on the biology of the parasite and to find the isoforms that provide the highest immunoprotection. At present, the identification of stage and tissue-specific FABP isoforms as well as their differential function in Fasciola spp. is still limited and the current study was performed to investigate differences in the expression patterns and immunogenicity of FABP isoforms from F. gigantica. It is expected that the obtained information advances the understanding of the biological roles of FABPs during the maturation of the parasite and that it will be useful for the development of a vaccine.
MATERIALS AND METHODS
Parasites
Unembryonated eggs and adult F. gigantica were collected from the gall bladders and bile ducts of naturally infected cattle killed at local slaughterhouses in Thailand. Intrauterine eggs were isolated from the uterus of freshly collected adults under a dissecting microscope. Metacercariae shed from experimentally infected snails (Lymnaea ollula) were used to orally infect ICR mice. Juvenile parasites were collected 2, 4, and 6 weeks after infection. Parasites were washed in 0·85% normal saline solution and kept in liquid nitrogen until required for further experiments.
Nucleic acid preparations
Genomic DNA was extracted from frozen adult F. gigantica as previously described (Grams et al. Reference Grams, Adisakwattana, Ritthisunthorn, Eursitthichai, Vichasri-Grams and Viyanant2006). Total RNA of eggs, juveniles and adults was extracted by using the TRIzol reagent (Invitrogen-Life Technologies, USA), according to the supplied protocol. The nucleic acids samples were quantified by spectrophotometry at 260 nm and were stored at −20°C until use.
Molecular cloning and sequence analysis
The isolation of FgFABP1 (GenBank Accession no. AF112568) has been described previously (Grams et al. Reference Grams, Vichasri-Grams, Sobhon, Upatham, Viyanant, Sirisinha, Chaiyaroj, Tapchaisri and 2000). A partial FgFABP3 cDNA was isolated by PCR from a metacercarial stage F. gigantica cDNA library using oligonucleotide primers based on F. hepatica FABP3 (GenBank Accession no. AJ250098). Subsequently, the partial cDNA was subcloned into the pGEM-T Easy vector (Promega, USA) and used as a DIG-labelled DNA probe (PCR DIG labeling Mix, Roche, Germany) to isolate a full-length FgFABP3 cDNA from the above library following standard screening procedures. The DNA sequences of both strands were determined by NSTDA Bioservice Unit, Thailand and by MWG AG Biotech, Germany. Nucleotide and deduced amino acid sequence analyses were performed with EMBOSS 2.8 (Rice et al. Reference Rice, Longden and Bleasby2000). NCBI-BLAST (Altschul et al. Reference Altschul, Gish, Miller, Myers and Lipman1990) through the NCBI server at http://www.ncbi.nlm.nih.gov/BLAST/ was used to search for and to retrieve homologous sequences. C. sinensis: Q8MUC1; E. granulosus: EgFABP1 Q02970, EgFABP2 Q9BMK3; F. gigantica FgFABP1 Q9UAS2; F. hepatica: FhFABP1 Q7M4G0, FhFABP2 Q7M4G1, FhFABP3 Q9U1G6; S. japonicum: Sj14 O45035, SjFABP2 AAW26894; S. mansoni: Sm14 AAM18480, SmFABP2 XP_002576002. Clustal X was used to create multiple alignments (Larkin et al. Reference Larkin, Blackshields, Brown, Chenna, McGettigan, McWilliam, Valentin, Wallace, Wilm, Lopez, Thompson, Gibson and Higgins2007).
Southern and Northern analysis
For Southern analysis, 10 μg each of genomic DNA were digested in separate reactions with restriction endonucleases Hind III, Xho I, and Hind III/Xho I (Fermentas Life Sciences, Lithuania). The reaction products were size-separated in 0·7% agarose gels in TBE buffer. Lambda DNA digested with EcoR I and Hind III was used as size standard. For Northern analysis, 30 μg total RNA from adult F. gigantica was heat-denatured in 50% formamide/2·2 m formaldehyde in 1×MOPS buffer before size separation in a 1·2% agarose gel containing 2·2 m formaldehyde in 1×MOPS buffer. A high-range RNA molecular weight marker (Invitrogen-Life Technologies) was used to determine the size of detected RNAs. Nucleic acids were transferred to Hybond-N Plus nylon membranes (Amersham Biosciences, UK) and immobilized by baking for 90 min at 80°C. Hybridization was performed at 55°C (DNA) or 68°C (RNA) in 50% formamide, 5×SSC, 0·02% SDS and 2% blocking reagent (Roche) for 16 h with digoxigenin-labelled DNA probes. The FgFABP1 and FgFABP3 DNA probes were synthesized using specific oligonucleotide primers and a PCR DIG labeling Mix (Roche). Enzymatic detection was done by using anti-DIG-alkaline phosphatase (Roche) and NBT/BCIP substrate (Fermentas Life Sciences).
Stage-specific detection of FgFABP transcripts
The extracted total RNA was treated with DNase I (Fermentas Life Sciences) at 37°C for 30 min and the enzyme inactivated by heating at 65°C for 10 min in the presence of 25 mm EDTA. Then 200 ng of DNase I treated total RNA was reverse transcribed by SuperScript III reverse transcriptase (Invitrogen Life Technologies) for 1 h at 55°C using the reverse primers listed below. The first strand cDNA was used as template to perform a standard PCR (35 cycles at 94°C, 57°C, 72°C, 1 min each step) with 3 primer pairs used in each reaction sample to amplify a 380 bp FgFABP1 fragment (FABP1RT-F: 5′-TTC GTG GAA GTA TGG CGA C-3′ and FABP1RT-R: 5′-TCA CGC TTT GAG CAG AGT G-3′), 299 bp FgFABP3 (FABP2RT-F: 5′-CTG ATC ACA AGT TCC AAA CC-3′ and FABP2RT-R: 5′-TTG TGG TAA TTG TTC ACA GC-3′) and 496 bp glutathione S-transferase, FgGST (GSTRT-F: 5′-AAC ATC TGT ACG GTC GTG AT-3′ and GSTRT-R: 5′-GAT TCC ATG TAT GCC TTG AT-3′). The RT-PCR products were size separated in a 1·8% agarose gel and GeneRuler 100 bp Plus DNA Ladder (Fermentas Life Sciences) was used as a size standard.
Construction of FgFABPs expression plasmids
A yeast expression system was used to express soluble rFgFABPs. The FgFABP cDNAs were subcloned into the YEpFLAG-1 vector (Kodak, USA) using terminal-located restriction endonucleases sites (lower case) introduced by PCR. FgFABP1 was inserted using 5′ Xho I and 3′ BamH I sites while FgFABP3 was inserted using 5′ Xho I and 3′ Sac II sites. In addition, the reverse primers introduced hexahistidine-tag (bold) and stop codon (italics). The following primers were used YFgFABP1-F: 5′-ctc gag GCT GAC TTT GTG GGT TCG-3′, YFgFABP1-R: 5′-gga tcc TCA GTG GTG GTG GTG GTG GTG CGC TTT GAG CAA GTG GT- 3′, YFgFABP3-F: 5′-ctc gag GCC AAT TTT GTG GGT TCG-3′, and YFgFABP3-R: 5′-ccg cgg TTA GTG GTG GTG GTG GTG GTG TAC TTT GTG GTA ATT GTT C-3′. After transformation of E. coli DH5α and plasmid preparation in vitro mutagenesis was performed by inverse PCR to remove a glycosylation site in FgFABP3. Codon 101 was changed from AAC (Asn) to TAC (Tyr) with the following oligonucleotide primers (the nucleotide substitution is indicated in lower case) Mut-F, 5′-GTT CAA AAG TGC CCG GAA tAC ACC-3′ and Mut-R, 5′-CTG GGA CAT TTT GCT GTC GGA ATC-3′. The change in the DNA sequence was confirmed by sequencing (Micromon, Monash University, Australia). YEpFLAG-1-FgFABP1, YEpFLAG-1-FgFABP3 and original YEpFLAG-1 plasmid DNA were used for transformation of S. cerevisiae BJ 3505 by facilitation with PEG-Bicine (Klebe et al. Reference Klebe, Harriss, Sharp and Douglas1983).
Preparation of recombinant FgFABPs from yeast cells and production of polyclonal anti-FgFABP antisera
Transformant yeast cells were selected by culture in minimal medium minus tryptophan. After incubation at 28°C for 3 days, positive colonies were picked and recovered onto the same medium. A starter culture was prepared by inoculation of single yeast colony into 15 ml of liquid medium adding uracil and L-lysine-HCl (Law et al. Reference Law, Smooker, Irving, Piedrafita, Ponting, Kennedy, Whisstock, Pike and Spithill2003). The culture was incubated at 28°C with shaking for 48 h, subsequently diluted 1:20 into YPHSM (1% dextrose, 3% glycerol, 1% yeast extract, 8% peptone and 20 mm CaCl2) and protein expression was then allowed for 72 h. The culture supernatant containing rFgFABP1 or rFgFABP3 was collected and mixed with buffer A to a final concentration of 50 mm NaH2PO4, 300 mm NaCl and 10 mm imidazole, pH 8·0. The insoluble material was pelleted by centrifugation at 5000 g for 20 min at 4°C and the supernatant fraction was loaded onto Ni-NTA affinity chromatography columns. The recombinant protein was purified under native conditions according to the QIAExpressionist manual (QIAGEN). After verification of purification by SDS-PAGE analysis the elution fractions containing rFgFABPs were pooled, dialysed against PBS, pH 7·2 at 4°C for 24 h, and stored at −20°C for further study.
ICR mice obtained from the National Laboratory Animal Centre, Mahidol University, Thailand were used to produce polyclonal rFgFABP antisera. The mice (n =6 for each antigen) were subcutaneously immunized with 15 μg rFgFABP1 or rFgFABP3 in TiterMax adjuvant (Sigma, USA) 3 times at 2-week intervals. The antisera were collected 1 week after the last immunization and stored together with the pre-immune sera at −20°C.
Parasite antigen preparations
Eggs released into the bile of infected cattle were isolated by sedimentation from the collected bile. They were repeatedly washed in PBS, pH 7·2 and observed under the microscope. To extract the soluble egg (SE) antigen the eggs were homogenized and sonicated (6 periods of 10 s with 10 s pauses at 200–300 W) in PBS, pH 7·2, containing 1 mm PMSF. The homogenate was centrifuged at 15 000 g for 30 min at 4°C to remove insoluble material and the supernatant was collected. Excretory/secretory (ES) product was prepared from freshly obtained adult parasites. The parasites were washed several times in 0·85% normal saline solution and then transferred into PBS, pH 7·2, containing 1× antibiotic-antimycotic (Invitrogen Life Technologies). The parasites were incubated at 37°C for 4 h. The supernatant containing ES product was collected after removing insoluble material by centrifugation at 15 000 g for 30 min at 4°C. Cirrus sac antigen (CSA) was prepared from cirrus sacs removed from fresh adult parasites under a microscope by using a needle. CS and Crude whole worm extract (CW) were prepared by tissue homogenization in lysis buffer (10 mm Tris–HCl, pH 7·2, 150 mm NaCl, 0·5% Triton-X 100, 1 mm EDTA, 2 mm PMSF). Insoluble material was pelleted by centrifugation at 15 000 g for 15 min at 4°C. The supernatant was collected and dialysed against PBS, pH 7·2. SE extract, ES product and CSA were concentrated using Microcon YM-3 centrifugal filter units (Millipore, Bedford, USA). Protein concentration was determined by a Bradford assay (Bio-Rad, USA) and all antigen preparations were stored at −20°C until use.
Sera of F. gigantica-infected rabbit and sheep
Two New Zealand white (NZW) rabbits were each infected orally with 200 F. gigantica metacercariae. Serum samples were collected pre-infection and bi-weekly post-infection for up to 12 weeks. The pooled sera of 10 sheep, each infected with 225 F. gigantica metacercariae, were kindly provided by David Piedrafita, Monash University, Australia. They were sampled pre-infection and bi-weekly post-infection for up to 10 weeks.
SDS-PAGE and Western blot
The parasite antigens (10 μg CW extract, SE extract, CS extract and 15 μg ES product) and rFgFABPs (200 ng) were size-separated in 15% SDS-PAGE gels. The gels were then either stained with Coomassie Blue (0·025% Coomassie Blue R-250, 40% methanol, and 7% acetic acid) or electrotransferred to Hybond™ ECL nitrocellulose membranes (Amersham Biosciences) by a wet blot apparatus (Mini Trans-Blot Electrophoretic Transfer Cell, Bio-Rad) at 100 V for 90 min. The blotted membranes were blocked with 5% skim milk in TBS, pH 7·5, containing 0·1% Tween 20 at room temperature for 90 min before incubation with diluted antisera (1:200) sampled from infected rabbit and sheep. The polyclonal antisera raised against rFgFABPs in mice were used as positive controls at a dilution of 1:500 to react with the membrane-bound parasite extracts. Alkaline phosphatase-conjugated goat anti-rabbit and goat anti-mouse immunoglobulins (Zymed, USA) and horseradish peroxidase-conjugated rabbit anti-sheep (DAKO) were used as secondary antibodies (1:2000). Enzymatic detection was done by using either NBT/BCIP substrates or SIGMAFAST™ 3,3′-diaminobenzidine with metal enhancers (Sigma).
Immunohistochemical detection of FgFABP1 and FgFABP3 in F. gigantica
Freshly obtained juvenile and adult F. gigantica were fixed in 4% paraformaldehyde in PBS, pH 7·4, and embedded in Paraplast following standard procedures. Sections were cut at 7 μm and mounted on Histogrip (Zymed)-coated slides. The deparaffinized and rehydrated sections were incubated in 10 mm citrate buffer, pH 6·0, and epitope retrieval was done 3×5 min in a microwave oven at 700 W. Endogenous peroxidase activity was eliminated by incubation with 3% H2O2 for 30 min. Non-specific binding sites were blocked by incubation in 1% glycine in TBS, pH 7·5, for 5 min and 4% BSA in TBST (TBS, pH 7·5 containing 0·1% Tween 20) for 90 min. Afterwards, the sections were incubated with mouse anti-rFgFABP1 or FgFABP3 antisera (1:800) in 1% BSA in TBST for 16 h. Biotinylated rabbit anti-mouse immunoglobulin (DAKO, Denmark) was used as secondary antibody (1:200) for 30 min. After washing in TBS, pH 7·5, the sections were incubated with avidin-biotin peroxidase (Vectastain ABC Kit, Vector Laboratories, Canada) for 30 min. Chromogenic detection was done using aminoethyl carbazole (AEC) substrate solution (Zymed) according to the manufacturer's instructions. Pre-immune and infected mouse sera (1:800) were used as negative and positive control, respectively. The analysis was performed entirely at room temperature.
2D gel electrophoresis, immunoblotting and mass spectrometry
CW extract, ES product, and SE extract were prepared by using a 2D Clean-up Kit (Amersham Biosciences). The prepared protein samples were then solubilized in rehydration buffer (8 m urea, 2% CHAPS, 50 mm DTT and 0·2% Bio-Lyte ampholytes) and 50 μg protein in a total volume of 125 μl were applied to 7 cm linear pH 3–10 IPG or pH 7–10 strips (Bio-Rad) by passive rehydration for 16 h at 20°C. In the first dimensional separation all strips were focused at 250 V, 4000 V and 15 000 V-h using the PROTEAN IEF cell (Bio-Rad). After focusing, the IPG strips were reduced in equilibration buffer (6 m urea, 0·375 m Tris-HCl, pH 8·8, 2% SDS and 20% glycerol) containing 2% (w/v) DTT (Bio Basic INC, Canada) for 15 min and then alkylated in equilibration buffer containing 2·5% iodoacetamide (Sigma) for 15 min. The separation by molecular mass was carried out on the Protean II system (Bio-Rad) in 15% polyacrylamide gels at 200 V until completion. The resulting gels were stained with Bio-Safe TM Coomassie Blue G-250 (Bio-Rad) or transferred to nitrocellulose membranes by semi-dry blotting using a Fastblot B33 instrument (Whatman Biometra, Germany) for 1 h 10 min at 60 mA. Immunodetection was achieved as described above for Western blot analysis. Detected protein spots were excised and analysed by mass spectrometry (LC/MS, Genome Institute, BIOTEC, NSTDA, Thailand) to identify the protein.
RESULTS
Molecular cloning and characterization of FgFABP cDNAs
The genome of Fasciola contains a gene family encoding fatty acid binding proteins (Espino and Hillyer, Reference Espino and Hillyer2001; Espino et al. Reference Espino, Rodriguez Medina and Hillyer2001). A cDNA encoding FgFAPB1 of F. gigantica (GenBank Accession no. AF112568) had been previously isolated (Smooker et al. Reference Smooker, Hickford, Vaiano and Spithill1997; Grams et al. Reference Grams, Vichasri-Grams, Sobhon, Upatham, Viyanant, Sirisinha, Chaiyaroj, Tapchaisri and 2000). The screen of a meta-cercarial stage F. gigantica cDNA library for a homologue of F. hepatica FhFABP3 (AJ250098) resulted in a full-length cDNA encoding FgFABP3. This cDNA has a total size of 544 bp, containing a 38 bp 5′ UTR, 399 bp ORF, 80 bp 3′ UTR, and 26 bp poly (A) tail. The nucleotide sequence data are accessible in the GenBank database under the Accession number GU479978. The FgFABP1 and FgFABP3 cDNAs encode proteins of 14·6 kDa molecular mass with 132 amino acid residues and their deduced amino acid sequences have 66·7% identity and 78% similarity in global pairwise comparison but quite different isoelectric points of 4·96 and 9·9, respectively. Remarkably, FgFABP3 is identical in its sequence to FhFABP3 (UniProtKB Accession Q9U1G6) even at the nucleic acid level and is so far the first fully conserved sequence of the two species that has been observed. Three additional RT-PCR amplifications of the FgFABP3 transcript were performed with separately extracted RNA from single adult parasites and the subsequent direct PCR product sequencing demonstrated that each worm contained the same 2 alleles different by 8 silent base exchanges, always at the third nucleotide position in a codon. High sequence identity/similarity values were also observed with homologous proteins from the trematodes S. japonicum (Sj14, 48·9%/68·4%), S. mansoni (Sm14, 48·5%/69·7%), and Clonorchis sinensis (47·4%/66·9%) and to a lesser degree with the cestode Echinococcus granulosus (EgFABP1, 37·3%/56·0%) and Homo sapiens (B-FABP (FABP7), 39·1%/57·9%) which was also evident in the multiple sequence alignment (Fig. 1).

Fig. 1. Multiple alignment of the deduced amino acid sequences of FgFABP1 and FgFABP3 with the amino acid sequences of FABPs from F. hepatica, S. japonicum, S. mansoni, C. sinensis, and E. granulosus. The sequences are sorted by decreasing identity values to FgFABP3 and their Accession numbers are listed in the Materials and Methods section. Residues identical to those in FgFABP3 are indicated by dots, similar residues are in lowercase, non-conserved residues are in uppercase, gaps are indicated by (-). Clustal X determined consensus residues are shown on top with (*) indicating fully conserved residues, colons indicating highly conserved, and dots indicating less-conserved residues. Lowercase letters ‘s’ and ‘h’ on top indicate strand and helical structures as determined for Sm14. The underlined N101 in FgFABP3 indicates the substituted residue in yeast recombinant FgFABP3 (N101Y).
Analyses of F. gigantica nucleic acids for FgFABP3 sequences
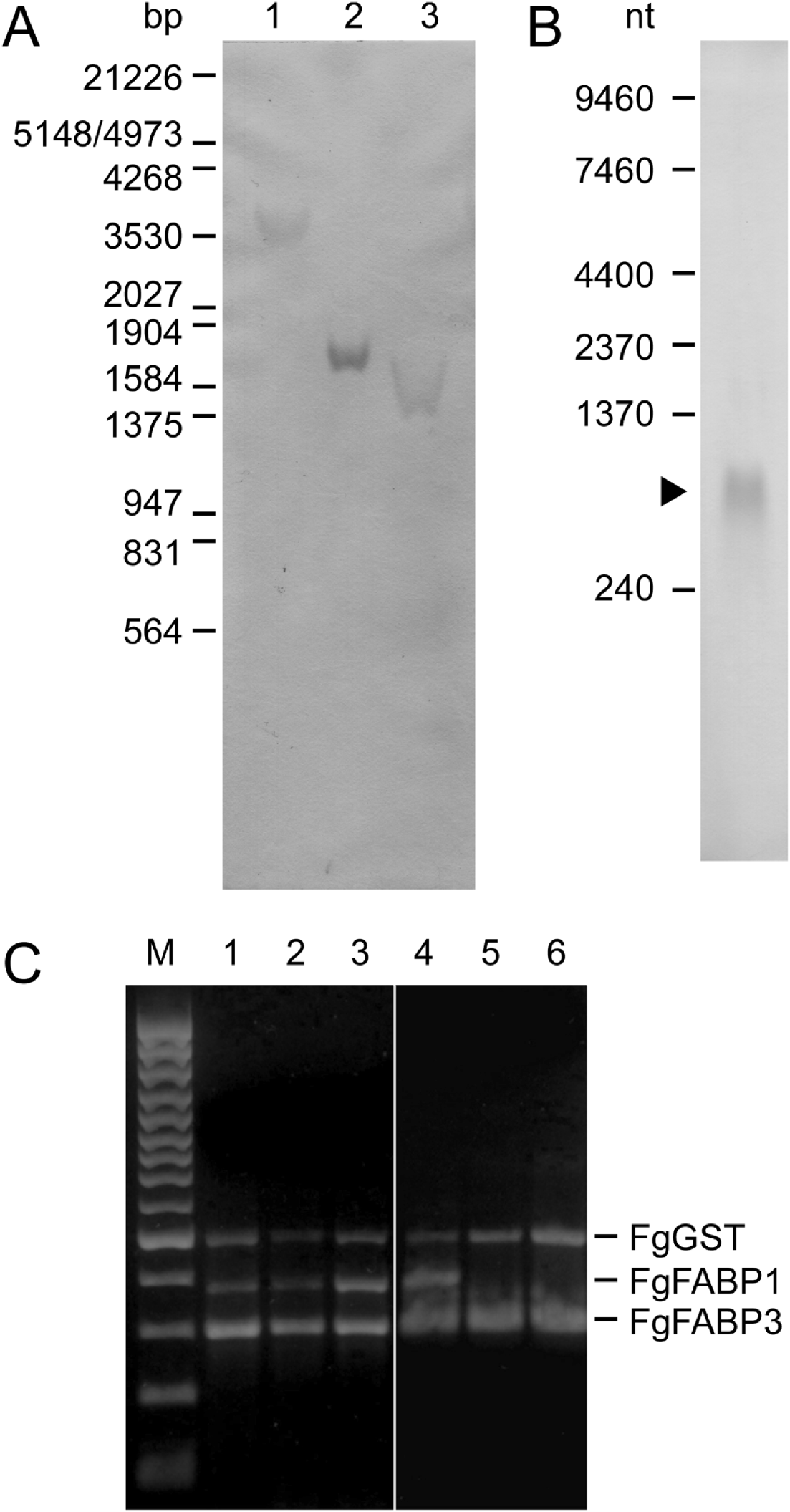
In order to confirm the presence of the isolated FgFABP3 sequence in the F. gigantica genome and to evaluate the FgFABP3 gene copy number, a Southern hybridization analysis was performed. Only a single hybridizing DNA fragment was detected in F. gigantica genomic DNA digested with either Hind III, Xho I or Hind III/Xho I using the FgFABP3 cDNA as a probe (Fig. 2A). The same probe was used in Northern analysis of total RNA from adult parasites and detected a 600 nucleotide transcript (Fig. 2B). The stage-specific presence of FgFABP RNA was investigated by RT-PCR on total RNA from 2-, 4-, 6-week-old juveniles and adult parasites with FgFABP gene-specific primers and demonstrated that FgFABP1 and FgFABP3 transcripts are present at all times. But only FgFABP3 transcript was found in intrauterine and bile-released eggs (Fig. 2C).
Fig. 2. Fasciola gigantica nucleic acid analyses by a DIG-labelled FgFABP3 DNA probe (A, B) and by reverse transcriptase PCR using FgFABP1, FgFABP3 and FgGST specific primers(C). (A) Southern hybridization of genomic DNA digested with restriction endonucleases Hind III (1), Xho I (2), and Hind III/Xho I (3). λ DNA digested with EcoR I/Hind III was used as size standard. (B) Northern hybridization of 30 μg total RNA from adult F. gigantica. Positions and fragment sizes of the RNA size standards are indicated at the left. (C) RT-PCR products obtained with gene-specific primers and 200 ng total RNA extracted from 2-week-old (1), 4-week-old (2), 6-week-old (3) juveniles, adult (4) F. gigantica, eggs released into the bile (5), and intrauterine eggs (6). The three gene-specific primer pairs were used together in all reaction samples. A 100 bp DNA ladder was used as size standard (M).
Production of recombinant FgFABPs and immune response to FgFABPs in infected rabbit and sheep
Soluble recombinant FgFABP1 and FgFABP3 (rFgFABP1 and rFgFABP3) were expressed in S. cerevisiae with an N-terminal FLAG epitope and a C-terminal hexahistidine-tag. The expression was monitored by SDS-PAGE and silver staining and also verified using an anti-histidine-tag antibody on membrane-bound protein. Recombinant FgFABP1 was abundantly expressed as a single protein with a relative mobility of M r 16 500. On the other hand, rFgFABP3 was expressed in 2 forms with relative mobilities of M r 16 500 and M r 23 500 as non-glycosylated and glycosylated protein respectively. Treatment with N-glycosidase F or PNGaseF removed this N-linked glycosylation from rFgFABP3 (data not shown). After removal of the glycosylation site in FgFABP3 by in vitro mutagenesis (N101Y) only non-glycosylated rFgFABP3 was obtained and used in all further studies. The immune response of F. gigantica-infected rabbit and sheep to FgFABP1 and FgFABP3 was analysed with immunoblots. The rabbit sera detected rFgFABP3 starting with serum sampled 6 weeks post-infection whereas rFgFABP1 was only detected starting 10 weeks post-infection (Fig. 3A). The sheep sera did not react with the recombinant proteins.

Fig. 3. Immunological detection of recombinant and native FgFABPs. (A) Immunoblotted rFgFABPs probed with sera of a rabbit infected with Fasciola gigantica metacercariae collected 0, 2, 4, 6, 8, 10, and 12 weeks post-infection. The grey arrowheads indicate the 15·5 kDa rFgFABPs products. (B) Immunoblotted F. gigantica 10 μg CW extract, 15 μg ES product, 10 μg SE antigen, 10 μg CS extract, and 200 ng rFgFABP1 and rFgFABP3 probed with mouse anti-rFgFABP1 and rFgFABP3 antisera.
Immunoblot analysis of rFgFABPs and F. gigantica antigen preparations using anti-rFgFABP1 and anti-rFgFABP3 antisera
Polyclonal anti-rFgFABP1 and anti-rFgFABP3 antisera were produced in mice by subcutaneous immunization with the purified recombinant protein for the immunodetection of recombinant and native proteins. The two anti-FgFABP antisera showed some cross-reactivity to the other rFgFABP isoform. Anti-rFgFABP1 readily detected a doublet of 15·7 and 14·7 kDa antigens in CW extract and less strongly an antigen doublet at 14·7 and 13·7 kDa in the ES product. Anti-rFgFABP3 detected single 14·6 kDa and 13·7 kDa antigens in CW extract and ES product, respectively. In addition, both antisera reacted with 13·7 kDa antigens in SE and CS extracts (Fig. 3B).
Immunolocalization of FgFABPs in tissue of adult and juvenile F. gigantica
The anti-rFgFABP antisera were further used to analyse the distribution of native FgFABPs in the tissue of juvenile and adult F. gigantica by immunohistochemistry. Both antisera strongly detected FgFABPs in the parenchymal tissue and also the oral and ventral suckers of the parasites in the two developmental stages. However, the staining in the parenchyma was heterogeneous with weak to strong intensity as previously reported (Pankao et al. Reference Pankao, Sirisriro, Grams, Vichasri-Grams, Meepool, Kangwanrangsan, Wanichanon, Ardseungneon, Viyanant, Upatham and Sobhon2006). A differential tissue-specific reactivity of the two antisera was observed in the intestinal epithelium and the reproductive tissues (Fig. 4). In the case of the anti-rFgFABP1 antiserum, weak staining was found in the intestinal epithelium, vitellaria, and intraoval vitelline cells while strong staining was found in the seminal vesicle, cirrus, testis, and also spermatozoa around the intrauterine eggs. In contrast, using anti-rFgFABP3 antiserum, a reversed staining intensity was observed for these tissues (Fig. 4).

Fig. 4. Immunolocalization of FgFABPs in the tissue of Fasciola gigantica using mouse anti-rFgFABP1 (3, 5, 7, 9, 11, 13, 15) and anti-rFgFABP3 (4, 6, 8, 10, 12, 14, 16) antisera. (1) Negative control, mouse pre-immune serum; (2) positive control, serum of F. gigantica-infected mouse; (1–4) sagittal sections in the area of the ventral sucker, anterior is to the left; (5, 6) cross-sections through the cirrus sac; (7, 8) high magnification of intrauterine eggs; (9, 10, 13, 14) cross-sections in the central area of the parasite; (11, 12) high magnification of testis tubules; (15, 16) high magnification of vitellaria. Abbreviations: Ca, caecum; Cs, cirrus sac; Eg, egg; Os, oral sucker; Pc, parenchyma; Sp, spermatozoa; Sv, seminal vesicle; Tt, testis tubule; Tg, tegument; Vi, vitellaria; Vs, ventral sucker.
2DE and mass-spectrometric identification of FgFABPs in antigen extracts
Antigen patterns of CW extract, ES product and SE extract of F. gigantica were resolved by 2DE using linear pH 3–10 IPG strips and controlled for quality by Coomassie Blue G staining (data not shown). The anti-FgFABP antisera were used to detect FgFABP isoforms in 2DE immunoblots of these extracts (Fig. 5). FgFABPs of ∼14 kDa molecular mass were detected with isoelectric points between 5 and 10. In CW extract at least 5 protein spots strongly reacted with each of the two anti-rFgFABP antisera, 3 spots migrating at lower pH displayed as a doublet of slightly different molecular weight proteins. In the ES product, only 2 spots were detected matching the predicted pI 5 of FgFABP1 and pI 10 of FgFABP3. Anti-rFgFABP1 reacted stronger with the spot at lower pH while anti-rFgFABP3 showed similar signal intensity with both spots. As in the CW extract, the spot at pH 5 was a doublet of proteins. In the SE extract the antisera reacted with 2 spots migrating at pH 8 and 10 with anti-rFgFABP1 resulting in a weaker signal than anti-rFgFABP3. Moreover, anti-rFgFABP3 antiserum faintly reacted with a third spot (Fig. 5). The detected spot at pH 10 in the SE extract corresponded to the predicted pI of FgFABP3 and in 2DE immunoblot of ES products and SE extract separated in the first dimension on linear pH 7–10 IPG strips anti-rFgFABP3 reacted stronger with this spot than with the other 2 spots (data not shown).

Fig. 5. 2DE-immunoblots of Fasciola gigantica CW extract ES product and SE antigen (50 μg each). Parasite extracts were separated across a linear pH range of 3–10 in the first dimension and 15% SDS-PAGE in the second dimension. The membrane-transferred proteins were probed with the indicated antisera. The negative control, mouse pre-immune serum, did not react with any antigens and is not shown. Only the membrane areas of the resolved 14 kDa antigens are shown. The spots indicated by numbers 1 to 5 were used for identification by mass spectrometry.
In order to identify whether the strongly reactive spots in the ES product and SE extract were FgFABP1 or FgFABP3, spots 1 and 2 (Fig. 5) reactive with anti-rFgFABP1 antiserum and spots 3 and 4 (Fig. 5) for ES product and SE extract reactive with anti-FgFABP3 antiserum were analysed by mass spectrometry. Spots 1 and 2 were identified as FgFABP1 (P=2.71E-09/1.25E-12) whereas spots 3 and 4 were identified as FhFABP3 (P=8.88E-12/5.91E-12). Spot 5 in the SE extract migrating at pI 8 could not be assigned to a known FABP isoform but the highest scoring match obtained was with FhFABP3 through the conserved motif LGINVIKR.
DISCUSSION
We have demonstrated that F. gigantica shows a differential tissue-specific expression of FABP isoforms and that these isoforms cause a differential immune response in infected rabbits. In detail we have analysed the FgFABP1 and FgFABP3 isoforms, which have 66·1% sequence identity. Remarkably, F. gigantica shares a fully conserved FABP3 isoform with F. hepatica and we have verified the sequence with repeated cDNA generation from independent RNA sources. The result suggests that high sequence conservation of Fasciola FABP3 is essential to its function and that this function is essential to the parasite genus. Sequence conservation within the cytosolic FABP family is, in general, low as is evident from the HMM sequence logo of the family seed alignment in the PFAM database at http://pfam.janelia.org/family/PF00061. Significant exceptions are the highly conserved N-terminal residues G6 and W8 that are also found in the presently known FABPs of Platyhelminthes. In comparison with S. mansoni Sm14 residues involved in binding of arachidonic acid (Angelucci et al. Reference Angelucci, Johnson, Baiocco, Miele, Brunori, Valle, Vigorosi, Troiani, Liberti, Cioli, Klinkert and Bellelli2004) are highly conserved (T74, D76, R78 (K78 in Fg/FhFABP1), Q96, T103, I105 (V105 in Fg/FhFABP3)) in all Fasciola FABPs but are also found, for example, in human M-FABP (FABP8). Richieri et al. (Reference Richieri, Ogata, Zimmerman, Veerkamp and Kleinfeld2000) reported that mammalian FABPs did not exhibit a high degree of selectivity for specific fatty acids but that orthologues might have conserved tissue-specific functions across species. While FgFABPs show the highest sequence similarities to human B-, M- and T-FABPs they and the other described platyhelmintic FABPs form a monophyletic clade in phylogenetic analysis (Esteves et al. Reference Esteves, Joseph, Paulino and Ehrlich1997), which suggests gene duplication within the phylum and subsequent adaptation to specific functions in the parasites.
How does the differential tissue distribution of FABPs in F. gigantica compare to other trematodes? Based on published genome data, schistosomes have a very limited set of cytosolic FABPs. While there are a number of slightly different FABP isoforms in the databases they are all allelic as we could only find 1 gene locus by TBLASTN searches with the Sm14 (AAM18480) sequence of the S. mansoni genomic DNA assembly version 3.1 at the Sanger Institute at http://www.sanger.ac.uk/cgi-bin/blast/submitblast/s_mansoni, and similarly for searches with the Sj14 (O45035) sequence using NCBI-TBLASTN and S. japonicum whole-genome shotgun reads. The genomic data confirm the Southern analysis of Scott et al. (Reference Scott, Kennedy and McManus2000) who suggested a single copy gene and the gene structure analysis of Ramos et al. (Reference Ramos, Figueredo, Pertinhez, Vilar, do Nascimento, Tendler, Raw, Spisni and Ho2003). Furthermore, these database queries revealed a second, more distantly related FABP (XP_002576002 53% similarity to Sm14, AAW26894 48·9% similarity to Sj14; SmFABP2 and SjFABP2) in both species which seems to also originate from a single copy gene and, as such, the investigation of function and distribution of cytosolic FABPs in schistosomes is at present incomplete. Moser et al. (Reference Moser, Tendler, Griffiths and Klinkert1991) reported Sm14 immunostaining in male S. mansoni as being present in tubercles, ‘structures traversing the muscle layers’, and ‘some staining in the body of the parasite’. Gobert et al. (Reference Gobert, Stenzel, Jones and McManus1997) showed sex-specific anti-Sj14 reactivity in S. japonicum in vitelline droplets in females, and in lipid droplets below the subtegumental region of males, and Brito et al. (Reference Brito, Oliveira, Oliveira, Street, Riengrojpitak, Wilson, Simpson and Correa-Oliveira2002), localized Sm14 to the basal lamellae of gut and tegument in male S. mansoni. While Fasciola is a hermaphrodite it is of interest to notice the partial sex-specific distribution of FgFABPs; FgFABP1 is found in the male reproductive tissue while FgFABP3 has a supportive role for the female reproductive system through its presence in vitelline cells. In addition, as others have already reported (Espino and Hillyer, Reference Espino and Hillyer2001) and as supported by our data, Fasciola has a larger set of FABP isoforms encoded by distinct genes. Our 2DE data demonstrate that there is at least 1 additional isoform present in the soluble egg antigens that could not be fully identified by mass spectrometry. The possibility of unknown juvenile-specific FgFABPs must also be considered. The detailed distribution of FgFABP1 among parenchymal cell types in the adult has already been reported by us (Pankao et al. Reference Pankao, Sirisriro, Grams, Vichasri-Grams, Meepool, Kangwanrangsan, Wanichanon, Ardseungneon, Viyanant, Upatham and Sobhon2006). From other trematode genera no data have been published in respect to tissue-specificity, e.g., Opisthorchis/Clonorchis, Paragonimus. In the cestode Echinococcus granulosus 2 conserved FABPs (75·2% identity) have been investigated with the proteins detected in the protoscolex (Esteves et al. Reference Esteves, Dallagiovanna and Ehrlich1993, Reference Esteves, Portillo and Ehrlich2003). In the case of EgFABP1 the tegumental level of the protoscoleces showed the strongest staining. No staining was detected in the germinal layer. As previously mentioned, mammals have a larger repertoire of FABP isoforms, each of which, while named by the first tissue it has been found in, is present in several tissues, e.g. L-FABP was co-localized with I-FABP in the small intestine of rats (Shields et al. Reference Shields, Bates, Bass, Best, Alpers and Ockner1986). Co-localization of FgFABP1 and FgFABP3 in the parenchyma does not necessarily indicate a redundant function. Angelucci et al. (Reference Angelucci, Johnson, Baiocco, Miele, Brunori, Valle, Vigorosi, Troiani, Liberti, Cioli, Klinkert and Bellelli2004) discussed the observed high ligand affinity of Sm14 at acidic pH in view of its localization to the gut/tegument basal lamellae and suggested that this protein would take up fatty acids carried across membranes and distribute/release them in cell compartments with higher pH. The authors contrasted this to the opposite behaviour of mammalian adipocyte FABP and related this also to the differences in the isoelectric points of the two FABPs that could, in the case of a high pI, facilitate better membrane contact. While Fasciola FABP3 has a high pI, with the side-chains of lysine residues facing outward (using a model based on the resolved structure of Sm14), and is located in the gut epithelium we observed uniform cytoplasmic staining and not a higher intensity towards cellular membranes. This was the case for both isoforms in all tissues and should be re-investigated in greater detail by electron microscopy. Investigations concerning the lipid distribution in adult F. gigantica demonstrated lipids not only in parenchymal cells but also around intestinal caeca and testis tubules (Pankao, Reference Pankao2006). Interestingly, the above-mentioned putative second FABP of schistosomes has a low pI and if Angelucci et al. (Reference Angelucci, Johnson, Baiocco, Miele, Brunori, Valle, Vigorosi, Troiani, Liberti, Cioli, Klinkert and Bellelli2004) are correct then this FABP and FgFABP1 are involved in non-membrane associated intracellular fatty acid binding, transport, and release.
With regard to immunogenicity, while FABP1 and FABP3 transcripts were found from the metacercarial stage onward FABP3 caused a much earlier immune response in an infected rabbit, 6 versus 12 weeks post-infection. The 12-week rabbit serum detected both isoforms in 2DE blots of the adult ES product. Immunization with soluble rFABPs demonstrated that both isoforms are immunogenic. In conclusion, FABP3 must be released from the worm early in development and as Espino et al. (Reference Espino, Rodriguez Medina and Hillyer2001) have demonstrated an immune response to FABPs as early as 2 weeks after infection, the release should begin immediately after infection and be continuous. Regarding the source tissue, based on our results FABP3 is most abundant in parenchymal cells, followed by vitelline cells (including in eggs) and the gastrodermal epithelium. Obviously, the latter is the most promising candidate as a source for released FABP as parenchymal cells have no direct connection to the host environment and the development of vitelline cells starts only later in development. We did not observe tegumental staining. In comparison, the hourly sampled ES product of adult parasites contained a lower amount of FABP than CW extract, indicating that the majority is not released. However, in soluble egg antigen FABP3 and a second isoform with pI 8 are abundant; both should originate from vitelline cells and could be accidentally released in the adult if vitelline cells, unused in egg formation, are passed through the uterine tract into the host environment. Similarly, FABP1, which was not detected in the intestinal epithelium but in spermatocytes/spermatozoa, could be released from later developmental stages carried inside these cells.
In 2004, Raina et al. reported the absence of a humoral immune response against F. gigantica FABP in infected sheep, cattle, and buffaloes using native and rFgFABP1 in ELISA and immunoblots. Morphew et al. (Reference Morphew, Wright, Lacourse, Woods and Brophy2007) could not detect FABP in the bile of infected sheep but could in the ES product of parasites cultured in vitro using mass-spectrometry for identification of proteins separated by 2DE. Increased amounts of FABP isoforms of low pI were observed only after prolonged incubation times (>8 h) and FABP3 was only released by dead parasites. We have confirmed the negative results for F. gigantica-infected sheep; their sera did not react to recombinant or native FABP in ELISA and immunoblots. This is in contrast to the results of Hillyer (Reference Hillyer1995) and Espino and co-workers (2001) and this study where differential immune responses against FABP isoforms were observed in mice and rabbits. Emphasis is on ‘differential’ immune responses, e.g. early and late which suggests that the isoforms are temporally presented to the immune system due to developmental availability and are not only presented by stressed/dying/dead parasites. Species differences in the responses could be due to differences in the immune system of mice and rabbits, a different behaviour of the parasites in these small animals causing an increased release of FABPs or any combination of the two. Interestingly, Lee and Yong (Reference Lee and Yong2004) reported cross-reactivity of human sera from patients infected by Clonorchis sinensis, F. hepatica, and Paragonimus westermani to recombinant C. sinensis FABP in immunoblots. This would support the findings in mice and rabbits and emphasize that parasite/host interaction is at least partially specific to the exact combination of antigen and species. Unfortunately, all vaccine trials that have been performed on recombinant protein have only tested FABP1 which is an isoform causing a late humoral immune response in mice and rabbits. Clearly, using as a vaccine an isoform which can naturally induce an immune response at an earlier stage of parasitism should lead to better protective responses. Additional trials in which other isoforms or combinations thereof are applied would allow new insights into the use of FABPs as a vaccine in fascioliasis.
ACKNOWLEDGEMENTS
We would like to thank Dr David Piedrafita, Monash University, Melbourne, Australia for providing the sera of F. gigantica-infected sheep used in this study.
FINANCIAL SUPPORT
This research was supported by grants from Mahidol and Thammasat Universities, Thailand and the Commission on Higher Education, Ministry of Education, Thailand.