


INTRODUCTION
All species of the pseudophyllidean tapeworm genus Diphyllobothrium infect fish-eating vertebrates, with 14 being causative agents of diphyllobothriasis in humans (Ashford and Crewe, Reference Ashford and Crewe2003). Of these, D. latum is the most geographically widespread, occurring in Northern Europe (Peduzzi and Boucher-Rodoni, Reference Peduzzi and Boucher-Rodoni2004), North America (Rausch and Hilliard, Reference Rausch and Hilliard1970), South America (Santos and de Faro, Reference Santos and de Faro2005), and possibly also in Asia, including Japan and Korea (Lee et al. Reference Lee, Chai, Hong, Sohn, Huh, Cheong and Kang1989; Yamane et al. Reference Yamane, Shiwaku, Fukushima, Isobe, Yoneyama, Qiang and Jie1996). It is also recognized as an emerging disease in some countries (e.g. in Brazil) (Sampaio et al. Reference Sampaio, de Andrade, Lucas Mda, Fung, Gagliardi, Santos, Mendes, Eduardo and Dick2005), as developments in mariculture, the world trade in fish and fish products, and the trend for humans to eat raw fish (e.g. see Macpherson, Reference Macpherson2005), increases the incidence of diphyllobothriasis. The movement of fish and fish products, and the multiple species of Diphyllobothrium causing diphyllobothriasis drive the need to rapidly and accurately identify the larval forms of cestodes to species. The identification of plerocercoids of Diphyllobothrium from marine and freshwater fishes, using morphology alone, is considered a task for experts (Anderson and Gibson, Reference Anderson and Gibson1989). Recently, molecular tools have provided some progress. For example, Yera et al. (Reference Yera, Estran, Delaunay, Gari-Toussaint, Dupouy-Camet and Marty2006) reported the use of 2 mitochondrial genes for identifying Diphyllobothrium nihonkaiense, and Skerikova et al. (Reference Skerikova, Brabec, Kuchta, Jimenez, Garcia and Scholz2006) assessed the validity of D. pacificum as a valid species using the sequences of the second internal transcribed spacer (ITS-2) of ribosomal DNA (rDNA). There is a need to develop reliable diagnostic tests for species of Diphyllobothrium. The mitochondrial genome provides a rich source of molecular markers for the specific identification of helminths and for investigating population genetic structures (e.g. see Hu et al. Reference Hu, Chilton and Gasser2004; Hu and Gasser, Reference Hu and Gasser2006; Le et al. Reference Le, van De, Blair, Sithithaworn and McManus2006).
To date, the complete mitochondrial genomes of 13 flatworm species (6 from cestodes and 7 from trematodes, respectively) have been published. The sequences determined for Taenia crassiceps (see Le et al. Reference Le, Blair, Agatsuma, Humair, Campbell, Iwagami, Littlewood, Peacock, Johnston, Bartley, Rollinson, Herniou, Zarlenga and Mcmanus2000), T. solium (see Nakao et al. Reference Nakao, Sako and Ito2003), T. asiatica (see Jeon et al. Reference Jeon, Lee, Kim, Hwang and Eom2005), Hymenolepis diminuta (see von Nickisch-Rosenegk et al. Reference von Nickisch-Rosenegk, Brown and Boore2001), Echinococcus multilocularis (see Nakao et al. Reference Nakao, Yokoyama, Sako, Fukunaga and Ito2002), and E. granulosus (see Le et al. Reference Le, Pearson, Blair, Dai, Zhang and McManus2002b) all represent the order Cyclophyllidea. Comparative mitogenomics has rapidly become established as an approach for differentiating species and host-specific strains of Echinococcus (reviewed by McManus, Reference McManus, Maule and Marks2006).
Only 2 orders of cestodes are known to include species that infect humans. Cyclophyllideans, including Taenia, Echinococcus and Hymenolepis and the Pseudophyllidea, with Diphyllobothrium as the commonest of 6 genera; Diplogonoporus, Ligula, Pyramicocephalus, Schistocephalus, and Spirometra are the other 5 (Ashford and Crewe, Reference Ashford and Crewe2003). Pseudophyllidea and Cyclophyllidea are very different orders, both in terms of morphology, habitat, life-cycle and life-history strategies (see Caira and Littlewood, Reference Caira, Littlewood and Levin2001). Each represents distinct lineages in the evolutionary history of the cestodes, with cyclophyllideans recognized as representing one of the most derived lineages (Hoberg et al. Reference Hoberg, Mariaux, Brooks, Littlewood and Bray2001; Olson et al. Reference Olson, Littlewood, Bray and Mariaux2001), and pseudophyllideans variously placed as a basal difossate lineage (morphology: Hoberg et al. Reference Hoberg, Mariaux, Brooks, Littlewood and Bray2001) or a polyphyletic difossate assemblage (molecular data: Olson et al. Reference Olson, Littlewood, Bray and Mariaux2001). A recent molecular study by Brabec et al. (Reference Brabec, Kuchta and Scholz2006) formally recognized and circumscribed 2 lineages of pseudophyllideans. The first, including the families Diphyllobothriidae and Cephalochlamyidae, giving rise to the ‘Diphyllobothriidea’, which was resolved as a basal lineage amongst the difossate tapeworm lineages, and the second, more derived clade, the ‘Bothriocephalidea’ including the remaining 4 pseudophyllidean families. Sequencing the mitochondrial genome of D. latum may also provide improved insights into the use of mitogenomics for resolving cestode relationships.
Metazoan mitochondrial genomes are typically circular DNA molecules, ranging from 14 kb to 18 kb in size and contain 37 genes: 13 protein-encoding genes (cox1-cox3, cytb, nad1-nad6, nad4L, atp6 and atp8), 2 ribosomal RNA genes (rrnS and rrnL), 22 transfer RNA (trn) genes (Boore, Reference Boore1999), but the atp8 gene is lacking from most nematode and flatworm species reported thus far (Le et al. Reference Le, Blair and McManus2002a; Kim et al. Reference Kim, Eom and Park2006). Due to the uniformity of gene content across the diverse metazoan groups, the mitochondrial DNAs (mtDNAs) have attracted attention as one of the most useful genetic markers employed both for phylogenetic studies and genome evolution in a variety of animal groups. Another idiosyncratic feature of metazoan mtDNA is to evolve more rapidly than nuclear genomes, and there is a varying degree of substitution rates in different gene loci (e.g. Brown et al. Reference Brown, Prager, Wang and Wilson1982). These characteristics meet well with attempts to resolve deep-branching phylogeny of the major groups of metazoans (Lavrov et al. Reference Lavrov, Brown and Boore2004; Lavrov and Lang, Reference Lavrov and Lang2005) and to employ useful genetic markers for many phylogenetic (biogeographical, molecular ecological) studies of taxa with relatively recent origins (Avise, Reference Avise2000). Accordingly, complete mitochondrial genome sequences have become increasingly popular among contemporary molecular ‘phylogeneticists’, and the number of genome sequences published has been increasing during the last decade. In the present study, we determined the complete mitochondrial genome sequence of D. latum as a basis for the future definition of strain and species-specific markers, and for assessing mitogenomics in resolving the interrelationships of cestodes.
MATERIALS AND METHODS
Sampling and molecular techniques
An adult worm of Diphyllobothrium latum was obtained from a 31-year-old Russian female patient after anthelmintic treatment. The specimen was stored in 70% ethanol prior to DNA extraction. Total genomic DNA of D. latum was extracted using a QIAamp tissue kit (Qiagen Co.), according to the manufacturer's instruction and used as a template DNA for PCR amplification. Initially, 5 small fragments of D. latum mtDNA, ranging in size from ∼360 to 670 bp, were PCR-amplified using their corresponding primer sets for each of 5 gene regions (cob [CytbF/CytbR], nad1 [PLND1-F/PLND1-R], cox1 [p-1F/p-1R], rrnL [PL16S-F/PL16S-R], and rrnS [PL12S-F/PL12S-R]; see Table 1). PCR reactions (50 μl volume) were performed in 10 mm Tris-HCl (pH 8·4), 50 mm KCl; 2·5 mm MgCl2; 200 μm of each dNTP; 100 pmol of each primer and 2·5 U Taq polymerase (TaKaRa, Japan) under the following cycling conditions: 1 cycle (94°C for 3 min), 35 cycles (94°C for 1 min, 50°C to 60°C for 30 sec, 72°C for 1 min 30 sec), and 1 cycle (72°C for 10 min). The sequences obtained from the 5 amplicons were then used to design D. latum-specific primer sets for long PCR (see Table 1 for details of the primers). Five overlapping fragments (ranging in size from 450 bp to 5·5 kb) and covering the entire mitochondrial genome of D. latum were amplified using the Expand Long Template PCR System (Roche, USA) under the following conditions: 1 cycle of initial denaturation (45 s at 94°C), 35 cycles of denaturation-primer annealing-elongation (10 s at 92°C, 30 s at 63°C, and 8 min at 68°C), and 1 cycle of the final extension (12 min at 72°C). A negative control (no template) was also included in every PCR run. The amplicons were separated on 1% agarose gels, excised and purified using a QIAquick PCR purification kit (Qiagen, Germany). Sequencing of the amplicons was performed in both directions by ‘primer walking’ using a Big Dye Terminator Cycle-Sequencing Kit (Applied Biosystems), according to the manufacturer's instructions. Overlapping fragments were assembled to complete the sequence of the mt genome.
Table 1. The sequences and their relative positions of the PCR primers used in the present study
(The binding sites of the primers correspond to the relative positions in the mtDNA of Diphyllobothrium latum. The IUPAC codes were used for R (A, G), Y (C, T), W (T, A), K (T, G), S (C, G), H (A, T, C) and D (A, T, G).)

* The sources for the primers cited from previous studies.
Gene annotation and phylogenetic analyses
With the aid of automatic organelle genome annotation program (DOGMA; Wyman et al. Reference Wyman, Jansen and Boore2004), 12 protein-coding and 2 rRNA genes of Diphyllobothrium latum were identified by sequence comparison with those of other flatworm mtDNAs. Putative secondary structures of 22 tRNA genes were identified using DOGMA or via the recognition of potential secondary structures and anticodon sequences. Secondary structures of non-coding regions were predicted using the RNAdraw program (Matzura and Wennborg, Reference Matzura and Wennborg1996). For phylogenetic analysis, 7 species of cestode (including D. latum) for which complete mitochondrial genome sequences are available were included. The mitochondrial genome sequences used are as follows: D. latum (DQ985706; this study), Echinococcus granulosus (NC_008075), E. multilocularis (NC_000928), Hymenolepis diminuta (NC_002767), Taenia asiatica (NC_004826), T. crassiceps (NC_002547) and T. solium (NC_004022). The amino acid sequences were inferred from the 12 protein-coding genes of D. latum using the codon translation table #9 of the GenBank, and their identities verified by database searches. Inferred amino acid sequences were then subjected to sequence alignment using the program ClustalX (Thompson et al. Reference Thompson, Gibson, Plewniak, Jeanmougin and Higgins1997). The alignment of gene sequences was performed using the following options: gap opening penalty=10, gap extension penalty=1·0 with a ‘delay divergent sequence’ setting of 30% using the BLOSUM similarity matrix. A conserved block of the concatenated alignment was selected employing the program Gblocks (Castresana, Reference Castresana2000). The most recent molecular phylogeny of flatworm mtDNAs found the Trematoda as a sister group to the Cestoda (Park et al., Reference Park, Kim, Kang, Kim, Eom and Littlewood2007). Thus, Fasciola hepatica (NC_002546) and Paragonimus westermani (NC_002354) were used as outgroups for all phylogenetic analyses in this study. Bayesian analysis was performed using MrBayes 3.1 (Huelsenbeck and Ronquist, Reference Huelsenbeck and Ronquist2001). Four Markov Chain Monte Carlo (MCMC) chains were run for 106 generations, sampled every 100 generations. Bayesian posterior probability values representing the percentage of samples recovering particular clades were estimated after the initial 1000 trees (the first 105 generations) were discarded. Phylogenetic analyses were conducted using the maximum parsimony (MP) and neighbour-joining (NJ) methods, employing the program PAUP* 4.0b10 (Swofford, Reference Swofford2002). The MP analysis was performed with the exhaustive search option. The confidence level for each branch was estimated by nonparametric bootstrap analysis with 1000 random replications using a heuristic search option. The maximum likelihood mapping method (Strimmer and von Haeseler, Reference Strimmer and von Haeseler1997) was conducted to assess the amount of phylogenetic structure in the amino acid dataset, and the maximum likelihood (ML) tree was reconstructed using the TREE-PUZZLE 5.2 program (Schmidt et al. Reference Schmidt, Strimmer, Vingron and von Haeseler2002) with the mtREV24 matrix (Adachi and Hasegawa, Reference Adachi and Hasegawa1996) as an evolution model for mitochondrial proteins.
RESULTS AND DISCUSSION
Long-PCR amplification of D. latum mtDNA
The long-PCR amplification using each D. latum mtDNA-specific primer sets consistently generated a single amplicon. Primer sets (amplicon sizes) were as follows: Dl/n-cob-F1 and Dl-ND1-R1 (4 kb); Dl-ND1-F1 and Dl-CO1-R (2 kb); Dl-CO1-F and Dl-16S-R (1·3 kb); Dl-16S-F and Dl-12S-R (450 bp); and Dl/n-12S-F1 and Dl/n-cob-R2 (5·5 kb) (see Table 1 for details). The sequence identity in overlapping regions for these 5 long-PCR fragments was verified by comparison with the sequences of the 5 initial shorter amplicons from cob, nad1, cox1, rrnL and rrnS. All of the sequences were assembled to obtain the complete sequence of the mitochondrial genome of D. latum.
Gene content and organization
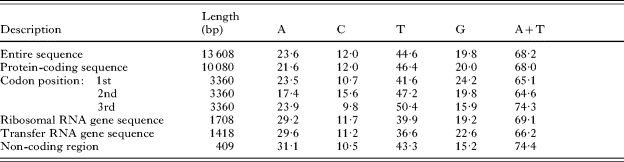
The complete mtDNA sequence determined for D. latum is 13 608 bp in length (GenBank Accession number; DQ985706), and it encodes 12 protein-coding genes (lacking atp8), 22 trn genes and 2 rRNA genes (Fig. 1), like other flatworm mtDNAs. All genes are transcribed in the same direction, a common feature of the flatworm mtDNAs reported thus far (Johnston, Reference Johnston, Maule and Marks2006; Littlewood et al. Reference Littlewood, Lockyer, Webster, Johnston and Le2006). The gene arrangement for the mitochondrial genomes of cestodes published to date, including that of D. latum, is the same, with the exception of that of Hymenolepis diminuta where the relative position of trnS2 and trnL1 is switched (von Nickisch-Rosenegk et al. Reference von Nickisch-Rosenegk, Brown and Boore2001). The relative position and length of each gene locus in the mt genome of D. latum are given in Table 2. The nucleotide composition of the entire mtDNA sequences is 23·6% A, 44·6% T, 19·8% G, and 12·0% C and is thus biased toward A and T (the A+T content of 68·2%; Table 3), similar to those of E. multilocularis (69%; Nakao et al. Reference Nakao, Yokoyama, Sako, Fukunaga and Ito2002) and E. granulosus (67%; Le et al. Reference Le, Pearson, Blair, Dai, Zhang and McManus2002b), but distinct from those of H. diminuta (71%; von Nickisch-Rosenegk et al. Reference von Nickisch-Rosenegk, Brown and Boore2001), T. crassiceps (74%; Le et al. Reference Le, Blair, Agatsuma, Humair, Campbell, Iwagami, Littlewood, Peacock, Johnston, Bartley, Rollinson, Herniou, Zarlenga and Mcmanus2000) and T. asiatica (71·4%; Jeon et al. Reference Jeon, Lee, Kim, Hwang and Eom2005).

Fig. 1. Circular representation of the mitochondrial genome of Diphyllobothrium latum. Genes are not drawn to scale, and the tRNA genes are indicated by hatched areas.
Table 2. Mitochondrial genome organization of Diphyllobothrium latum

Table 3. Nucleotide composition of the mitochondrial genome of Diphyllobothrium latum
Protein-coding genes
It is widely acknowledged that the nucleotide composition of protein-coding genes of metazoan mtDNAs is not randomly distributed (Saccone et al. Reference Saccone, Gissi, Reyes, Larizza, Sbisà and Pesole2002). This compositional bias is often associated with the amino acid sequence composition and unequal usage of synonymous codons within amino acid families (Herbeck and Novembre, Reference Herbeck and Novembre2003). For the protein-coding genes of D. latum mtDNA, amino acids inferred from T-, G-, and A-rich codons are very abundant: T-rich codons with more than 2 Ts in a triplet represent Phe (10·18% TTT and 1·37% TTC), Leu (8·00% TTA, 3·18% TTG, and 2·08% CTT), Ile (4·43% ATT), Val (4·61% GTT), Ser (3·27% TCT), Tyr (4·67% TAT) and Cys (2·56% TGT). The proportion of G-rich (more than 2 Gs in a triplet) and A-rich codons (more than 2 As in a triplet) are 12·36% and 9·75%, respectively, whereas C-rich codons account for 4·07% (Table 4). These 3 classes of codons, abundant for a specific nucleotide, represent 66·46% of all amino acids. The codon usage bias avoiding C is also particularly prominent at the third position of synonymous codons (Scouras and Smith, Reference Scouras and Smith2001). In almost all cases, the frequency of a specific codon is decreased when the third codon position is replaced with a C: The relative frequencies of Phe are 10·18% for TTT and 1·37% for TTC. The relative frequencies for Val are 4·61% for GTT, 2·11% for GTA, 2·14% for GTG, and 0·86% for GTC. The start and termination codons for the 12 protein-coding genes were inferred by comparing their sequences with those inferred from previously published mtDNAs of flatworm species. The 12 protein-coding genes of D. latum mtDNA were translated using the flatworm mitochondrial genetic code (translation table #9 in GenBank). All open reading frames (11 out of 12) are inferred to use ATG as an initiation codon, with the exception of cox3 for which GTG is used as the start codon. All 12 protein-coding genes are predicted to end with complete codons. Eight of 12 genes (cox3, nad4, atp6, nad2, nad1, nad3, cox1 and nad6) are inferred to terminate with TAG, whereas the others (cob, nad4L, cox2, and nad5) have TAA (see Table 2).
Table 4. Codon usage for 12 protein-coding genes of the Diphyllobothrium latum mitochondrial genome

* Stop (termination) codon.
Ab, Abbreviation.
Transfer RNA and ribosomal RNA genes
Twenty-two nucleotide segments, ranging in size from 56 to 71 nucleotides are predicted to fold into clover leaf-like secondary structures (Fig. 2), which are almost the same as those reported thus far for other cestode species. An amino-acyl stem of 7 nucleotide pairs and an anticodon stem of 5 nucleotide pairs are the common characteristics of 22 tRNAs found in D. latum mtDNA. Of 22 inferred tRNA secondary structures, 19 display the typical clover leaf-like configuration with a dihydrouridine (DHU)-arm, but this arm is lacking for the other 3 (trnS1, trnS2, and trnR). The D. latum trnC possesses a DHU arm as found in Hymenolepis diminuta and some other trematodes (Fasciola hepatica and Paragonimus westermani), but it is missing from other cestodes characterized to date (i.e. Taenia solium, T. asiatica, Echinococcus granulosus and E. multilocularis) and from all 5 schistosome species published thus far (see details in Littlewood et al. Reference Littlewood, Lockyer, Webster, Johnston and Le2006). The feature of trnC lacking a DHU arm, which is shared among some of cyclophyllidean cestodes and trematode groups, is likely to reflect an independent, homoplastic trait as it is absent from D. latum and H. diminuta (see Fig. 3). Based on sequence comparison with those of other neodermatan groups, 2 rRNA genes were identified: rrnL (968 bp) and rrnS (740 bp) are separated by the trnC (Table 1), and this gene arrangement is common to all other cestode and trematode mtDNAs reported thus far, but this is not the case for some monogenean mitochondrial genomes (Park et al., Reference Park, Kim, Kang, Kim, Eom and Littlewood2007).

Fig. 2. Predicted secondary structures of the 22 mitochondrial transfer RNAs of Diphyllobothrium latum.

Fig. 3. Phylogenetic relationships among eucestode species based on inferred amino acid sequence data selected from 12 mitochondrial protein-coding gene loci for 9 flatworm species. The topology of the trees constructed using different analytic approaches (MP, NJ, Bayesian, and ML methods) was the same. The numbers above the branches represent bootstrap percentages for maximum parsimony (MP), neighbour joining (NJ), posterior probability values for Bayesian phylogeny (BP), and the quartet puzzling supporting values for maximum likelihood (ML), respectively.
Non-coding regions
Eighteen intergenic sequence regions, representing a total length of 484 bp and varying from 1 to 222 bp, were detected in the mtDNA genome of D. latum. Of these, 2 non-coding regions (NR1 and NR2) were particularly prominent. Located between trnY and trnL1 (222 bp for NR1) and between nad5 and trnG (187 bp for NR2), respectively, these non-coding regions are still relatively small compared with those in trematodes (e.g. Littlewood et al. Reference Littlewood, Lockyer, Webster, Johnston and Le2006). In some schistosomes, however, the long non-coding region (conventionally called ‘LNR’) is A+T-rich and generally known to range in size up to 5–7 kb, showing the considerable length variation among strains (Le et al. Reference Le, Humair, Blair, Agatsuma, Littlewood and McManus2001). The A+T contents of the NR1 and NR2 of D. latum mtDNA are 77·0% (35·1% for A, 41·9% for T, 11·7% for G, and 11·3% for C) and 71·1% (26·2% for A, 44·9% for T, 19·3% for G, and 9·6% for C), respectively; the value for the NR1 is considerably higher than the average of the entire sequence (A+T content of 68·2%). Hairpin-like secondary structures of these non-coding regions were predicted. The secondary structures of 2 non-coding regions were inferred to contain 2 (NR1) and 4 (NR2) stem-loop structures, respectively (Fig. 4A and B). A hairpin-like stem-loop secondary structure is often found in the non-coding regions of cestode mtDNAs, but its functional role is not yet clear (cf. von Nickisch-Rosenegk et al. Reference von Nickisch-Rosenegk, Brown and Boore2001; Littlewood et al. Reference Littlewood, Lockyer, Webster, Johnston and Le2006).

Fig. 4. Secondary structures predicted for the non-coding regions identified in the mtDNA of Diphyllobothrium latum. The two non-coding regions NR1 (A) and NR2 (B) are located between trnY and trnL1 and between nad5 and trnG, respectively.
Mitochondrial molecular phylogeny of eucestodes
To assess the phylogenetic position of D. latum and the utility of mt genomes in resolving the interrelationships of cestode orders, an analysis of amino acid sequence data representing all 12 mitochondrial protein-coding gene loci for 9 selected flatworm species (including D. latum) was performed. A concatenated alignment set of 2980 homologous amino acid positions from conserved blocks was used. Based on maximum likelihood mapping analysis, more than 99·9% of all random samples of the quartet (33·3%, 28·6% and 38·0% in each trapezoid) were fully resolved. Of the 2980 homologous positions, 1280 variable sites were phylogenetically informative under the MP criterion. The exhaustive search option of the MP method yielded a single tree (length=5024 steps, CI=0·850, RI=0·645; Fig. 3). Phylogenetic relationships among the eucestodes using different analytical approaches (MP, Bayesian, NJ, and ML methods) are the same in their topology (see Fig. 3). Phylogenetic relationships among species are well resolved with maximal nodal support throughout. Monophyly of Taenia, Echinococcus (the family Taeniidae and the order Cyclophyllidea) is well supported. Hymenolepis diminuta, a member of the family Hymenolepididae was resolved as a sister taxon to the Taeniidae. This mitogenomic prediction is concordant with morphology-based phylogenetic hypothesis (Brooks et al. Reference Brooks, Hoberg and Weekes1991; Hoberg et al. Reference Hoberg, Mariaux, Justine, Brooks and Weekes1997, Reference Hoberg, Gardner and Campbell1999) and also with molecular estimates of eucestode phylogeny based on nuclear rDNA sequences (Mariaux, Reference Mariaux1998; Olson et al. Reference Olson, Littlewood, Bray and Mariaux2001). This information suggests that mitogenomic data sets provide a useful means of resolving cestode interrelationships. The traditional works for cestode classification have recognized dichotomous grouping within the class Cestodaria, represented by the monozoic orders Gyrocotylidea and Amphilinidea, and Eucestoda (Cestoidea sensuEhlers, Reference Ehlers1985), comprising all other cestode groups. Although there is little doubt about the monophyly of Eucestoda, its internal phylogeny, particularly phylogenetic relationships among major ordinal groups have not yet been fully resolved (Mariaux, Reference Mariaux1996; Hoberg et al. Reference Hoberg, Mariaux, Justine, Brooks and Weekes1997; Littlewood et al. Reference Littlewood, Cribb, Olson and Bray2001). In the present study, each of the genes is readily alignable, and there is considerable sequence variation between Diphyllobothrium and the cyclophyllidean mt genes available for phylogenetic analysis (with over 40% of the alignable sites being phylogenetically informative under parsimony). A recent study (Hardman and Hardman, Reference Hardman and Hardman2006) suggests that most of the amino acid sequences inferred from neodermatan mt genes performed well in resolving relationships among taxa for which mt genomes were available, with nad2 being most useful and nad5 being of least value. These authors concluded that a minimum of 4 kb of sequence (whether concatenated, or 40 samples of randomly selected 100 bp fragments) was needed to resolve expected nodes. Thus, further mitochondrial genome information from a broad range of major eucestode groups, including primitive orders (e.g. Caryophyllidea and Spathebothriidea) will likely provide a deeper insight into outstanding issues concerning cestode evolution, such as the origins and radiation of scolex morphology, patterns of strobilation and proglottization (e.g. see Olson et al. Reference Olson, Littlewood, Bray and Mariaux2001). Considering the relative ease with which entire genomes can now be characterized, we suggest sequencing additional exemplar taxa from other cestode orders, in particular the basal lineages, in order to select optimum gene regions and develop PCR-based protocols for mitochondrial DNA-based estimates of cestode phylogeny.
As revealed in earlier mitochondrial genome surveys (von Nickisch-Rosenegk et al. Reference von Nickisch-Rosenegk, Brown and Boore2001; Le et al. Reference Le, Pearson, Blair, Dai, Zhang and McManus2002b; Nakao et al. Reference Nakao, Yokoyama, Sako, Fukunaga and Ito2002; Jeon et al. Reference Jeon, Lee, Kim, Hwang and Eom2005), the gene arrangement of cestode mtDNAs is conserved, based on current information, and the pseudophyllidean D. latum mtDNA displays the same arrangement. The only exception is Hymenolepis diminuta for which the relative position of 2 tRNA genes (trnS2-trnL1) is switched, compared with those reported in all other eucestode studied to date. It is also anticipated that further mitochondrial genome surveys will improve our understanding of mitochondrial genome evolution, particularly of gene arrangements within the context of the phylogeny of major eucestode groups. Additional mitochondrial genome sequences for species of Diphyllobothrium will allow the identification of conserved gene regions for the design of PCR primers flanking regions of high variability for species and/or ‘strain’ identification. Of particular interest will be genes and non-coding regions offering higher rates of mutation for investigating population variation.
An adult worm of D. latum was provided by Dr Floriane de Marval (Laboratory of Unilabs Geneva, Switzerland). We thank the editor Robin Gasser and two anonymous reviewers for constructive comments and suggestions on an earlier version of the manuscript. DNA samples used in the present study were deposited in the Parasite Resource Bank of Korea National Research Resource Center, Republic of Korea. This work was supported by a Korea Research Foundation Grant (KRF-2003-015-C00445) to J. K. P.
