


INTRODUCTION
From the advent of single-gene amplification reactions in the late 1980s, and until not very long ago, many systematists have been content in fashioning phylogenetic trees for their group of interest from DNA sequence data on the basis of one or a few loci. Our motivations were, and largely still remain, centered on uncovering the specifics of the evolutionary relationships of the constituent higher taxonomic groups down to species so as to better describe their history of diversification. These self-styled ‘tree of life’ research programmes have borne considerable fruit, particularly in the last decade (e.g. Giribet, Reference Giribet2008; Hackett et al. Reference Hackett, Kimball, Reddy, Bowie, Braun, Braun, Chojnowski, Cox, Han, Harshman, Huddleston, Marks, Miglia, Moore, Sheldon, Steadman, Witt and Yuri2008; McLaughlin et al. Reference McLaughlin, Hibbett, Lutzoni, Spatafora’ and Vilgalys2009), during which time at-the-bench research efforts and costs associated with generating data have dwindled even as the scope of loci and numbers of taxa has accelerated. Similarly, the difficulties associated with such problems as sequence alignment and optimal tree discovery have been ameliorated considerably through computational advances driven by an intersection of evolutionary biology and the computer sciences (e.g. Bader et al. Reference Bader, Roshan and Stamatakis2006; Warren et al. Reference Warren, Sutton, Jones and Holt2007; Hittinger et al. Reference Hittinger, Johnston, Tossberg and Rokas2010; Kristensen et al. Reference Kristensen, Kannan, Coleman, Wolf, Sorokin, Koonin and Mushegian2010). Presently, the ease with which even model-based phylogenetic trees can be acquired for hundreds of terminals is such that the operationalism associated with the endeavour has progressively migrated from the rarefied realm of advanced research laboratories to being readily available to undergraduate and even secondary school instruction for basic biology course work (Kvist et al. Reference Kvist, Montanari, Yi, Fuks and Siddall2011).
Our collective success in taking what has been a research goal and transforming that into a prerequisite for research is reminiscent of similar transformations associated with earlier technological advances such as electron microscopy. That is, while the 1960s and 1970s marked an ‘age of discovery’ for the sub-cellular organization of Apicomplexa and their complex development, or the surface architecture of cestode worms, electron microscopy is now but a tool in comparative biology, not a research nexus in and of itself. The same must now be admitted as it pertains to molecular and morphological phylogenetic systematics. We believe that it is appropriate for some introspection regarding how the powerful tool of a phylogeny, at one time the consequential end-point, is being re-imagined in a broader context of a progressive research programme that continues to deepen our understanding of the natural world. In short, knowing the name of the song, what the song is called, and even what the name-of-the-song is called, each belie what the song actually is (Carroll, Reference Carroll1871).
Imre Lakatos aptly contrasted progressive and degenerative research programmes; the latter being marked by entrenchment, the former by its pursuit of novelties both of method and prediction (Lakatos, Reference Lakatos1971). Exemplary of the distinction between these two paradigms is embodied in the (socio-scientific) history of the pursuit of historical biogeography in a phylogenetic context. Early considerations of the spatiotemporal diversification of some clades consisted of little more than superimposition of phylogeny and cartography (Croizat, Reference Croizat1958). The idea was sufficiently novel to spur ever increasingly thoughtful methods of inference, some operationalist, others statistical, with a trajectory that has seen and continues to see advances like Component Analysis (Page, Reference Page1990), Biogeographic Parsimony Analysis (Brooks, Reference Brooks1990), Dispersal Vicariance Analysis (Ronquist, Reference Ronquist1997), and most recently LaGrange (Ree and Smith, Reference Ree and Smith2008) and BayesDIVA (Nylander et al. Reference Nylander, Olsson, Alström and Sanmartín2008). This field of inquiry clearly continues to satisfy the progressivism imagined by Lakatos (Reference Lakatos1971), even as the tendency to entrench oneself in one method or another might not be. Our intent is not to cast aspersions, having been as guilty of Croizatian generalized-arm-waving (Borda et al. Reference Borda, Oceguera-Figueroa and Siddall2008) as we have equally availed ourselves of more progressive considerations regarding the historical biogeography of leeches (Borda and Siddall, Reference Borda and Siddall2011). Rather, we hope to stimulate a deeper consideration of how the results of phylogenetic analyses (i.e. trees) might be brought to bear on the field of comparative biology in manner that already is technologically and conceptually well within reach; though perhaps ways underexploited by our field, systematics.
Using the Hirudinida, leeches, as a framework - historically notorious and yet an inexplicably understudied group of (mostly) ectoparasitic annelid worms – our aim here is to go ‘beyond the tree’. This is not to minimize the efforts several have made in the last 15 years to generate hypotheses of phylogenetic relationship for leeches; indeed, those efforts are necessary and central prerequisites to the discovery operations of historical correlates we might now explore. Instead, and drawing on each of microbiology, co-speciation, evolutionary selection and genomic evolution, we hope to characterize and describe a progressive research programme for a clade of charismatic microfauna in a way that inspires our hirudinological colleagues as much as it might have others give greater consideration to what can be accomplished with a tree-in-hand.
THE TREE AS A PREMISE
Underpinning any contemporary approach to comparative biology is a phylogenetic tree, or more typically a constellation of trees pertaining to the group of interest. With respect to leeches, work towards understanding their place among Annelida more generally, and of the various family, genus and species level relationships more specifically, began first with morphological (Siddall and Burreson, Reference Siddall and Burreson1995) and, quickly on the heels of that, molecular phylogenetic analyses (Siddall and Burreson, Reference Siddall and Burreson1998). Among the earliest discoveries stemming from this work were that leeches do not deserve their own Class-level status as Hirudinea, on a par with Oligochaeta and Polychaeta, but in contrast are simply a highly specialized group of oligochaete worms closely related to the Lumbriculida (Siddall et al. Reference Siddall, Apakupakul, Burreson, Coates, Erséus, Gelder, Källersjö and Trapido-Rosenthal2001). While perhaps an unwelcome diminishment of the taxonomic stature of the group, the findings are fortuitous for any long-term attempts at understanding their development, biochemistry or other evolutionary-associated phenomena. Suppose, for example, that dinosaurs were sister to crocodiles and birds, no extant data from crocodiles and birds could convincingly shed light on the unknowable characteristics of dinosaurian physiology or soft anatomy. The discovery that dinosaurs are arranged as a paraphyletic grade between crocodilians and birds (relegating Aves to a mere subset of theropods), allows for a more convincing understanding of ancestral states. So too with the clitellate annelids. Determination of ancestral states, even as it might concern gene families associated with blood-feeding, can now proceed by examination of the utility and complexity of homolgous loci in the lumbriculids and other groups of oligochaetes.
Leeches remain monophyletic in all analyses of the group. The phylogenetic relationships of various leech groups remains a work in progress, but one that has already revealed considerable information about within and among group relationships. Apakupakul et al. (Reference Apakupakul, Siddall and Burreson1999) remains the touchstone from which all other leech phylogenetic work derives. In that analysis we demonstrated the basic organization of leech evolutionary history with the early divergence of Glossiphoniidae, Ozobranchidae and Piscicolidae and confirming the sister group relationship of the hirudiniform and erpobdelliform leeches. Within those basic outlines, most of the suborders and families of Hirudinida have since been subject to phylogenetic scrutiny on the basis of molecular and morphological data including Glossiphoniidae (Siddall et al. Reference Siddall, Budinoff and Borda2005), Piscicolidae (Utevsky and Trontelj, Reference Utevsky and Trontelj2004; Williams and Burreson, Reference Williams and Burreson2006), predaceous Erpobdelliformes (Siddall, Reference Siddall2002; Oceguera-Figueroa et al. Reference Oceguera-Figueroa, Phillips, Pacheco-Chaves, Reeves and Siddall2011), and the Hirudiniformes of blood-feeding infamy (Borda and Siddall, Reference Borda and Siddall2004; Phillips and Siddall, Reference Phillips and Siddall2005; Borda et al. Reference Borda, Oceguera-Figueroa and Siddall2008; Phillips and Siddall, Reference Phillips and Siddall2009; Phillips et al. Reference Phillips, Arauco-Brown, Oceguera-Figueroa, Gomez, Beltran, Lai and Siddall2010). It is with respect to the latter that most of the rest of this contribution is concerned. That is, our recent research into phylogenetic correlates, both microbial and salivary, has focused on leeches once thought to comprise the Hirudinidae. These are vermiform, freshwater, swimming leeches with muscular jaws armed with denticles for cutting into flesh so as to allow the acquisition of a blood-meal. Fig. 1 amalgamates the current state of knowledge regarding leech phylogeny. As it pertains to the traditional composition of Hirudinidae, those taxa now are variously spread across the hirudiniforms in the families Hirudinidae, Macrobdellidae and Praobdellidae. Borda et al. (Reference Borda, Oceguera-Figueroa and Siddall2008) detailed the intermediate phylogenetic position of terrestrial leech families Haemadipsidae and Xerobdellidae between the New World medicinal leeches, Macrobdellidae, and Old World Medicinal leeches, Hirudinidae sensu stricto. Furthermore, various non-blood-feeding groups have been found to place among the ‘medicinal’ leech taxa (Phillips and Siddall, Reference Phillips and Siddall2009), and an entirely mammalophilic family, Praobdellidae, has been recently recognized as phylogenetically distinct from the other two ‘medicinal’ leech clades (Phillips et al. Reference Phillips, Arauco-Brown, Oceguera-Figueroa, Gomez, Beltran, Lai and Siddall2010).

Fig. 1. Composite metaphylogeny of the order Hirudinida based on a collection of prior work illustrating current knowledge of the relationships of most leech families. The relationships of the Hirudindiformes (below Hirudo verbana) indicate the complex evolutionary history of the ‘medicinal’ leech families Hirudinidae, Praobdellidae and Macrobdellidae. Each terminal represents a species in a molecular phylogenetic analysis. Branches are proportional to change within families. Backbone phylogeny based on Apakupakul et al. (Reference Apakupakul, Siddall and Burreson1999), Siddall et al. (Reference Siddall, Apakupakul, Burreson, Coates, Erséus, Gelder, Källersjö and Trapido-Rosenthal2001) and Phillips and Siddall (Reference Phillips and Siddall2009). Blood-feeding lineages in red; non-sanguivorous lineages in blue.
MICROBIAL CORRELATES
Like many other blood-feeding animals, leeches harbour select prokaryotic flora in association with their digestive tracts. Proboscis-bearing sanguivorous species possess mycetomal organs specific to this task with intracellular alphaproteobacteria or gammaproteobacteria (Kikuchi and Fukatsu, Reference Kiskuchi and Fukatsu2002; Siddall et al. Reference Siddall, Perkins and Desser2004; Perkins et al. Reference Perkins, Budinoff and Siddall2005). In contrast, jaw-bearing medicinal leeches in the Hirudinidae and Macrobdellidae host a limited flora in the intraluminal fluid of the crop (Graf, Reference Graf1999; Siddall et al. Reference Siddall, Trontelj, Utevsky, Nkamany and Macdonald2007a; Laufer et al. Reference Laufer, Siddall and Graf2008). To date, only a single culturable bacterial species has been detected in any individual medicinal leech: Aeromonas veronii in the European Hirudo verbana (Graf, Reference Graf1999, Reference Graf2002), which was often mistakenly reported as Hirudo medicinalis (Siddall et al. Reference Siddall, Worthen, Johnson and Graf2007b), Aeromonas jandaei in the North American Macrobdella decora (Siddall et al. Reference Siddall, Trontelj, Utevsky, Nkamany and Macdonald2007a), and either of these two Aeromonas species (but never both) in the crop of the European Hirudo orientalis (Laufer et al. Reference Laufer, Siddall and Graf2008). In addition to these individual culturable gammaproteobacteria, Worthen et al. (Reference Worthen, Gode and Graf2006) demonstrated the co-presence of an unculturable Bacteroidetes microbe closely related to Rikenella species in H. verbana.
The crop, or gastric caeca, occupies approximately one-third of a leech's body somites allowing the annelid to expand more than six times its unfed body weight during feeding, permitting extended periods between feeding events (Munro et al. Reference Munro, Siddall, Desser and Sawyer1992). The role of the resident microbial flora is not yet well elucidated. Functions could range from the provision of essential nutrients not readily available in a diet that is limited exclusively to blood (e.g. Nogge, Reference Nogge1981) to antimicrobial activities inhibiting putrefaction of the blood meal (Rio et al. Reference Rio, Anderegg and Graf2007).
Species of Aeromonas, including A. veronii and A. jandaei, are ubiquitous in circumglobal freshwater habitats raising questions regarding the historical maintenance of a single species of the genus in any given leech. Graf (Reference Graf2000) was first to suggest that oral vertical transmission is responsible insofar as all leeches must withdraw their oral anterior through the egg-bearing coccon after it is secreted by the clitellum. Corroborating this, the medicinal use of leeches has repeatedly demonstrated their propensity for introducing Aeromonas infections at a bite wound (Whitaker et al. Reference Whitaker, Kamya, Azzopardi, Graf, Kon and Lineaweaver2009). Recent work confirms that Aeromonas veronii is present as soon as H. verbana coccons are deposited (Rio, Reference Rio2008). The Rikenella-like symbiont is detectable later (Rio et al. Reference Rio2008). While not a prerequisite, such vertical transmission of associated microbes hints at emergent co-evolutionary histories (Moran, Reference Moran2001).
The revision of medicinal leeches into several families (Phillips et al. Reference Phillips, Arauco-Brown, Oceguera-Figueroa, Gomez, Beltran, Lai and Siddall2010) demonstrates that the two genera examined thus far for their intraluminal microbial crop symbionts are distantly related representatives of Macrobdellidae and the revised Hirudinidae (Fig. 1). Here we investigate the crop flora of a broader range of leech genera and families, and evaluate historical patterns of this tripartite symbiotic system.
Methodology
Intraluminal blood-meal was removed following transverse bisection of leeches at the region of the gastric tissue and well-anterior of the intestinal tract. DNeasy Tissue Kit (Qiagen Valencia, CA) was used for tissue lysis and DNA purification. Aeromonas-specific primers for DNA gyrase B (gyrB) were AerogyrBf TGTTGCTGACCATTCGTCGTAAC and AerogyrBr TTGGCATCGCTCGGGTTTTC with a predicted optimal annealing temperature of 59·4°C. Amplification reactions employed Taq Gold (Applied Biosystems) and 50 cycles of 94°C (45 sec), 55°C (45 sec) and 72°C (60 sec) following a 10 min pre-melt at 94°C. Bacteroidetes-specific primers employed for amplification of 16S rDNA from the co-symbiont and to avoid co-amplification of the gammaproteobacterium were SSUrik416F GCAGGAAGACGGCTCTATGAGTTG and SSUrik781 RATCGTTTACGGCGTGGACTACC with a predicted optimal annealing temperature of 56·7°C. Amplification reactions employed Ready-To-Go PCR Beads (GE Healthcare) and 35 cycles of 94°C (15 sec), 50°C (15 sec) and 72°C (40 sec) following a 4 min pre-melt at 94°C. PCR amplification products were purified with AMPure™ (Agencourt Bioscience Corporation). Cycle sequencing reactions were performed with an Eppendorf Mastercycler® using 1 μl Big Dye™ Extender Buffer v3.1, 1 μl of 1 μM primer and 3 μl of cleaned PCR template (13 μl total volume) and analyzed with an ABI PRISM® 3730 sequencer (Applied Biosystems). CodonCode Aligner (CodonCode Corporation) was used to edit and reconcile sequences. Sequences employed for comparative purposes were downloaded from NCBI. Alignments were accomplished using the European Bioinformatics Institute server for MUSCLE v. 3.7. Parsimony analyses were conducted in TNT v 1.1 (Goloboff et al. Reference Goloboff, Farris and Nixon2008) using ten replicates of random taxon addition, sectorial searching, the Ratchet (Nixon, Reference Nixon1999), and tree-fusing algorithms, with a requirement that the minimum length be found at least three times. Trees resulting from these new technology searches were submitted to tree-bisection-reconnection branch swapping retaining up to 10 000 trees. Resampling in TNT employed the parsimony jack-knife (Farris et al. Reference Farris, Albert, Källersjö, Lipscomb and Kluge1996), with five replicates of random taxon addition, sectorial searching, the Ratchet (Nixon, Reference Nixon1999), and tree fusing, with no requirement that the minimum length be found multiple times.
Data
Parsimony analysis of gyrB sequences (Fig. 2) for species of Aeromonas resulted in 100 equally parsimonious trees with 2232 steps for 504 informative characters and a retention index of 0·75. Each species of Aeromonas for which multiple sequences were available was resolved as monophyletic with jack-knife frequency values ranging from 67% for Aeromonas bestiarum towards 100% for most species. Isolates from European H. verbana and H. orientalis, Mexican Limnobdella mexicana and Southeast Asian Hirudinaria manillensis grouped in the A. veronii clade. Isolates from European H. medicinalis and African Asiaticobdella fenestrata grouped in the Aeromonas hydrophila clade. Isolates from North American M. decora and European H. orientalis grouped in the A. jandaei clade. Amplification reactions of gyrB were not successful with Asian Limnatis paluda and Hirudo nipponia, African Asiaticobdella buntonensis, or Australian Goddardobdella elegans.

Fig. 2. Consensus of 100 equally parsimonious trees resulting from analysis of gyrB sequences of species of Aeromonas as well as those isolated from the crop of hirudiniform leeches (bold). Asterisks denote sequences obtained from type-strains for species. Numbers at nodes are jack-knife frequencies (not shown within species). Relationships supported in fewer than 50 jack-knife replicates are represented by interrupted lines.
Parsimony analyses of the 16S rDNA sequences obtained from Bacteroidetes symbionts only of hirudiniform leeches (Fig. 3) resulted in 1 tree of length 321 for 115 informative characters and a retention index of 0·92. The resulting consensus grouped isolates in a manner that was highly congruent with the phylogenetic history of ‘medicinal’ leeches. That is, six of eight vertices of the 16S rDNA tree map to nodes on the leech tree without conflict (Fig. 3). The two conflicting vertices resulted from non-monophyly of the isolates from Asiaticobdella species and from the pre-exisiting isolate from H. verbana not grouping with other European Hirudo species.
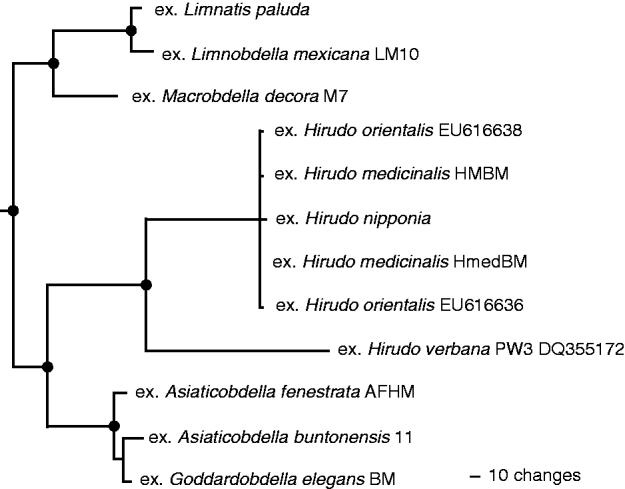
Fig. 3. Consensus of 105 equally parsimonious trees resulting from analysis of Bacteroidetes-specific 16S rDNA amplicons from hirudiniform leeches. Closed circles at nodes correspond to divergences that are consistent with leech phylogeny in Fig. 1.
Parsimony analysis of 16S rDNA sequences for a broader sampling of Bacteroidetes (Fig. 4) resulted in 36 equally parsimonious trees with 10 560 steps for 870 informative characters and a retention index of 0·67. Isolates from European and Asian species of Hirudo formed a clade sister to a fish gut symbiont, which together were more closely related to species of Alistipes than to Rikenella microfusus. The isolate from North American M. decora clustered nearby among a variety of uncultured and unidentified isolates, but closest to a termite gut symbiont. Isolates from African species of Asiaticobdella and the Australian G. elegans formed a clade deriving from among Pedobacter species. Isolates from the Asian L. paluda and the Mexican L. mexicana formed a clade deriving from among a paraphyletic assemblage of Flexibacter species.

Fig. 4. Consensus of 36 equally parsimonious trees resulting from analysis of 16S rDNA sequences of a variety of species and strains of Bacteroidetes, as well as those isolated from the crop of hirudiniform leeches (bold). Asterisks denote sequences obtained from type-strains for species. Numbers at nodes are jack-knife frequencies. Relationships supported in fewer than 50 jack-knife replicates are represented by interrupted lines.
The two distinct microbial groups resident in the intraluminal crop fluid of hirudiniform leeches exhibit markedly different patterns of historical conservation with respect to their leech hosts. In terms of the genus Aeromonas, and even though there appears to be marked host specificity and vertical transmission (Rio et al. Reference Rio, Maltz, McCormick, Reiss and Graf2009), leech phylogenetic relationships are entirely non-predicitive of the associations; nor is geography. That is, the North American M. decora (Macrobdellidae) and European H. orientalis (Hirudinidae) each harbour A. jandaei. Similarly, European H. medicinalis harbours the same symbiont, A. hydrophila, as the African A. fenestrata and is thus distinct from its European congeners H. verbana and H. orientalis. Aeromonas veronii proved to be the most ubiquitous culturable symbiont, inhabiting the crop of leeches in three distinct genera from two families across three continents. Whether or not species of Aeromonas are involved in the origins or maintenance of species-level cohesion where recently derived leech congeners are sympatric is an intriguing possibility.
The culturable gut flora of the European medicinal leech, originally named Pseudomonas hirudinis Busing, 1953, was widely considered to be a strain of A. hydrophila and has been a matter of some concern in the post-operative use of commercially available leeches (Whitaker et al. Reference Whitaker, Kamya, Azzopardi, Graf, Kon and Lineaweaver2009). Graf (Reference Graf1999) demonstrated that all isolates from commercially available leeches were actually A. veronii and that previous identifications as A. hydrophila were misled by the vagaries of chemotaxonomy. Since then, Siddall et al. (Reference Siddall, Trontelj, Utevsky, Nkamany and Macdonald2007a) have demonstrated that commercially available European medicinal leeches actually are H. verbana, not H. medicinalis. It is as ironic to discover H. medicinalis harbouring A. hydrophila, as it is accidentally fortuitous that H. verbana has been the leech commercially available for clinical use; it harbours a considerably less-pathogenic A. veronii (Silver et al. Reference Silver, Rabinowitz, Küffer and Graf2007).
Bacteroidetes symbionts, unlike species of Aeromonas, exhibit a considerably tighter historical association with their respective hosts, and one that is more obviously phylogenetically than geographically constrained. The phylogenetic results of Bacteroidetes leech symbionts alone (Fig. 3), while topologically remarkably similar to historical expectations from hirudiniform phylogeny (Fig. 1) prove illusory when reconsidered in the context of Bacteriodetes more fully (Fig. 4). Half of the apparent co-evolutionary pattern depicted in the leech-symbiont-only tree evaporates under broader phylogenetic consideration in a manner that should serve as a caution to other investigations of host-symbiont co-speciation. This problem of scale in co-speciation studies has a precendent in Nishiguchi et al.'s (1998) work concerning light-emitting symbiotic vibrionids in sepiolid squid. That is, while a preliminary analysis based on only seven species evidenced tight co-evolutionary patterns between Vibrio fischeri strains and squid hosts, an error of scale that is frequently taken for granted (Kimbell et al. Reference Kimbell, McFall-Ngai and Roderick2002; Kimbell and McFall-Ngai, Reference Kimbell and McFall-Ngai2003; Nishiguchi, Reference Nishiguchi2002; Nichiguchi et al. Reference Nishiguchi, Lopez and Boletzky2004; Soto, Reference Soto, Gutierrez, Remmenga and Nishiguchi2009), that co-speciation pattern was erased under fuller consideration of squid and fish associated strains of bioluminescent vibrionids (Dunlap et al. Reference Dunlap, Ast, Kimura, Fukui, Yoshino and Endo2007; Keading et al. Reference Keading, Ast, Pearce, Urbanczyk, Kimura, Endo, Nakamura and Dunlap2007).
In the broader evaluation of Bacteroidetes, however, the symbiont-leech associations retain more phylogenetic than geographic constraint (Fig. 4). All symbionts from species of Hirudo, whether European or Asian (H. nipponia) form a clade of apparently indistinguishable Alistipes species. Reflecting leech genus-level diversificiation (Fig. 1), and notwithstanding their inhabiting well-separated continents, Australian species of Goddardobdella are host to a symbiont that is closely related to (and barely distinguishable from) a Pedobacter species found in two African Asiaticobdella species. Similarly, symbionts of leeches in the Praobdellidae form a clade of Flexibacter-like species despite the obvious geographic separation of Mexico and Afghanistan. Taken together these results are suggestive of recent, but not ancient, tight historical association between leech hosts and their unculturable Bacteroidetes symbionts. A similar recent historical pattern has been noted in relation to glossiphoniid leeches and their mycetomal symbionts in which three clades of leeches, the genera Placobdella, Placobdelloides and Haementeria, are host to three distinct clades of endosymbiotic bacteria each occupying three distinct mycetomal morphological types (Perkins et al. Reference Perkins, Budinoff and Siddall2005).
Hints regarding a physiological role that the unculturable symbionts may play in the guts of their leech hosts comes from other Bacteroidetes symbionts of invertebrates. Flavobacteriaceae symbionts of termites, for example, are involved both in synthesizing essential amino acids and in recycling nitrogen from uric acid (Bourtzis and Miller, Reference Bourtzis and Miller2006). Whereas we were unable to amplify a Bacteroidetes 16S rDNA from the Asian H. manillensis, given its belonging to the family Hirudinidae (Fig. 1), we would anticipate such an isolate to group with others in Pedobacter. Likewise, others in the mammalophilic clade of mucous membrane feeders (i.e. the Praobdellidae) should prove to be relatively closely related to the marine Flexibacter flexis. The inconsistency with which we were able to amplify Aeromonas species relative to Bacteroidetes reflects the crop dynamics reported for these two symbionts; both flourish in response to a blood-meal but the unculturable Bacteroidetes symbiont persists at higher levels for considerably longer periods after feeding (Kikuchi and Graf, Reference Kikuchi and Graf2007).
EVOLUTION OF SALIVARY PEPTIDES
Leeches have a long and storied history in medicine. Most of that seems to have been misguided optimism pertaining to the balancing of humors in the face of various ailments (Jackson, Reference Jackson2001). Prior even to Hippocrates describing the utility of leeches in phlebotomy, the practice of leech-mediated blood-letting already was already central in Ayurvedic and other oriental medical practices (Sawyer, Reference Sawyer1999). Hardly the medieval European practice it is perceived to be, leeching reached a nexus under Napoleon's surgeon Broussais in the 19th century being considered safer than the raw and uncontrolled methods of venesection for phlebotomy that otherwise prevailed (Jackson, Reference Jackson2001). No one, it seems, has ever died from a leech bite. With the advent of clinical medicine, in perhaps the first controlled trials ever attempted, P.C.A. Lewis demonstrated the futility of leeching as applied to prognoses associated with pnuemonitis and pleurisy (Moraiba, Reference Morabia1996). Notwithstanding the dubious utility of leeches for the treatment of obesity, hysteria and other ailments in the 19th century, the European medicinal leech, Hirudo medicinalis, since then has come to play a valuable role in the postoperative treatment of venous congestion following flap and replantation surgery (Derganc and Zdravic, Reference Derganc and Zdravic1960; Batchelor et al. Reference Batchelor, Davison and Sully1984). Leeches were recently approved as a medical device by the US-FDA (Rados, Reference Rados2004) and the anticoagulant property of leech saliva remains the subject of some considerable scientific scrutiny. The first successful attempt at human clinical dialysis treatment, for example, was only made possible through Haas (Reference Haas1924) employing the anticoagulative properties of a newly purified hirudin from European medicinal leeches. It appears that this was the first use of an animal-derived compound for clinical purposes. The use of hirudin was quickly superceded by porcine-derived heparin, yet hirudin has remained a leech-derived protein of considerable interest; particularly in cases of heparin-induced thrombocytopenia (HIT) (Greinacher et al. Reference Greinacher, Völpel, Janssens, Hach-Wunderle, Kemkes-Matthes, Eichler, Mueller-Velten and Pötzsch1999).
Different species of even closely related leeches exhibit known variation in the anticoagulant cocktail (Min et al. Reference Min, Sarkar and Siddall2010). Evolution's own site-directed mutagenesis has determined the components necessary for a compound to function successfully. Bioactive compounds that are expressed as gene products have ‘passed the test’ of evolutionary selection, unlike much of the results of in vitro structure-activity relationships. Phylogenetic examination of site-by-site values of the relative rates of non-synonymous to synonymous substitution over deep evolutionary time can identify negatively and positively selected sites in a peptide, thus permitting the identification of functional domains in otherwise too-large antigenic molecules.
The development, characterization and structure of novel therapeutic agents and molecular diagnostic tools from living organisms is an active area of biomedical research (hundreds of published papers in the last 5 years alone just for leeches). A determination of the molecular variation of salivary bioactive peptides is critical to the development of new therapies and tools for haematology. Besides hirudin, a variety of bioactive compounds already has been isolated (typically with HPLC and peptide sequencing) from the salivary secretions of Hirudo and Haementeria. These include (Baskova and Zavalova, Reference Baskova and Zavalova2001; Salzet, Reference Salzet2001): the original angiogenesis-inhibiting antistasin, as well as orthologues such as ghilanten and bdellastasin, each serine-protease (factor Xa) inhibiting antistasins with potent anti-metastatic abilities; the fibrinogenolytic hementin; bdellin, a non-classical Kazal-type plasmin-trypsin inhibitor; eglin, a leucocyte/mast cell elastase inhibitor; orgelase, a heparanase-like endoglucuronidase; destabilase, which promotes the dissolution of polymerized fibrin; and calin, which, unlike the preceding protease inhibitors, acts by blocking vWF-mediated binding of platelets to collagen glycoproteins. Already, some of these bioactive compounds are in pre-clinical development or in clinical trials for drug delivery and glaucoma (orgelase), emphysema and inflammation (eglin), or reduction of tumour metastases (bdellastasin).
Hirudo medicinalis and Haementeria ghilianii are only distantly related (Fig. 1), having diverged evolutionarily about 200 million years ago. Hirudo feeds by making a cutaneous incision whereas Haementeria inserts a muscular proboscis. Hirudo species are restricted to Europe where they feed on frogs, fish and only occasionally mammals. Haementeria species are confined to New World Tropics where the Giant Amazonian leech specializes on anacondas, crocodilians and the plentiful aquatic mammalian fauna like capybaras. Different species of medicinal leech (Hirudo sp. and their close allies) are already known to have evolved to produce distinct suites of bioactive compounds in their salivary secretions (Min et al. Reference Min, Sarkar and Siddall2010); for example, the North American medicinal leech is unique in secreting a 39 amino-acid peptide, decorsin, inhibiting platelet aggregation by blocking membrane glycoprotein IIb-IIIa integrins. Thus, the far more distantly related Giant Amazonian leech is certain to have even more radically diverged and potentially valuable components in its salivary secretions. We anticipate that hementin, which appears unique in its ability to promote the dissolution of platelet-rich clots, is just the first example.
At present, little is known regarding the genomic organization of these peptides, their copy number or to what degree orthologous loci are distributed across the various kinds of blood-feeding leeches. The available leech genome from Helobdella robusta (from the Joint Genome Institute) sheds little light on these questions, because this species is a predator on aquatic invertebrates rather than feeding on blood. It is surprising then to discover, through screening the annotated H. robusta genome via JGI's portal, three loci orthologous to antistasin (i.e. scaffold_ 49:464509-465641, scaffold_49:517938-518879, scaffold_49:1286706-1287330), one of which is actually expressed in Helobdella robusta embryos (CAXA11664 corresponds to scaffold_49:1286706-1287330), and a tandem array of six copies of leech antiplatelet protein (LAPP) on Helro1 scaffold 2, five of which are expressed in the living organism (CAWZ13451, CAXA9903, CAWZ1735, CAWZ1685, CAWZ7874). As expected, however, none of the other previously characterized leech bioactive salivary peptides appear in the Helobdella robusta genome draft.
The evolutionary history of leech salivary peptides associated with blood-feeding is only just beginning to be revealed. Faria et al. (Reference Faria, Junqueira-de-Azevedo Ide, Ho, Sampaio and Chudzinski-Tavassi2005) generated the first salivary Expressed Sequence Tag (EST) library from a leech; specifically Haementeria depressa. In that library of comparatively few (898) clones they only found ESTs homologous to LAPP, tridegin, and therostasin. In contrast, with over 2,000 transcripts we found a much wider array of bioactive proteins from the salivary glands of Macrobdella decora (Min et al. Reference Min, Sarkar and Siddall2010). These included the antiplatelet proteins saratin and decorsin, protease inhibitors like antistasins, eglin, bdellin and hirudin, the fibrinolytic destabilase, and an endoglucuronidase. All but decorsin had previously only been known from other leech species. In addition, Min et al. (Reference Min, Sarkar and Siddall2010) noted lectoxins, ficolins and histidine-rich proteins among the most frequent transcripts, raising the possibility that leeches have even more biomedically interesting secretions than previously thought.
With the same techniques as were detailed in Min et al. (Reference Min, Sarkar and Siddall2010), we have now successfully generated additional EST libraries from the European Hirudo verbana and from the African Asiaticobdella fenestrata (Fig. 1). Even the choice of these taxa was driven by the phylogenetic premise in Fig. 1 so as to include a broad array of medicinal leeches both in terms of known phylogenetic diversity as well as geographic diversity. From that work it appears that the platelet disintegrin decorsin is unique to the Macrobdellidae, but that each of hirudin, destabilase, antistasins, saratin, eglin and bdellin predate the origin of the various medicinal leech families.
Because this work comes at a time when we have well-corroborated evolutionary trees for medicinal leech phylogeny (Fig. 1), a variety of analytical approaches can enrich our understanding of functional and phenotypic constraints on these various protease inhibitors and other anticoagulants. It is now well established that simple pairwise comparisons of orthologous peptide sequences is insufficient (Rocha et al. Reference Rocha, Smith, Hurst, Holden, Cooper, Smith and Feil2005) for proper detection of rates of non-synonymous to synonymous substitutions (dN/dS or ω). The HyPhy statistical phylogenetic computing package (Kosakovsky Pond et al. Reference Kosakovsky Pond, Frost and Muse.2005) provides three possibilities for evaluating positive selection (ω >> 1·0) and negative selection (ω << 1·0) for whole molecules as well as for residue-level information. Amino acid residues that are under negative (or purifying) selection exhibit fewer amino acid changes than expected and, thus, may be critical to the historical functioning of an anticoagulant (useful for active site prediction). Whole peptides exhibiting overall more amino acid changes than expected are under positive selection as would be anticipated in evolutionary arms-race scenarios (Kosiol et al. Reference Kosiol, Vinar, da Fonseca, Hubisz, Bustamante, Nielsen and Siepel2008).
Implementation of the PARRIS method (Scheffler et al. Reference Scheffler, Martin and Seoighe2006) in HyPhy entails codon-based likelihood ratio tests on whole sequences even where the history of a protein is confounded by domain-shuffling recombination; a potentially confounding phenomenon in the history of leech antistasins (Mason et al. Reference Mason, McIlroy and Shain2004). While robust, the foregoing is best suited to examination of whole transcripts. In contrast, Kosakovsky Pond et al. (2005 - and implemented in HyPhy) have found that for relatively small data-sets, an implementation of fixed effects likelihood (FEL) models can accurately identify individual amino acid residues that deviate significantly from neutral evolution expectations. Moreover, the FEL method, and a Bayesian random effects (REL) approach overcome the lack of statistical power inherent in simple counting strategies (Pond and Frost, Reference Pond and Frost2005). Both FEL and REL determinations take into account topological relationships and relative branch lengths for orthologues and their hypothesized ancestral sequences. A more computationally intensive (yet ultimately tractable) module in HyPhy employs Bayesian evolutionary network graphical modeling to identify pairs (and higher-order combinations) of residues that have changed in concert across time. This mapping of residue-residue interactions accurately predicts tertiary structural adjacency for amino acid sites operating in concert or compensatory changes in adjacent residues (Poon et al. Reference Poon, Lewis, Kosakovsky Pond and Frost2007).
For the orthologous copies of hirudin already obtained from our EST libraries, and adding to this the orthologous hirullin and haeamdin (Fig. 5), these apparently single-copy transcripts sort out phylogenetically in a manner that exactly mirrors that expected from higher level relationships (Fig. 1). Hirudin binds irreversibly to the fibrinogen binding exosite of thrombin as well as to the catalytic active site pocket. With an inhibition constant in the picomolar range, it remains the most potent natural direct thrombin inhibitor (DTI) known (Greinacher and Warkentin, Reference Greinacher and Warkentin2008). Moreover, hirudin, unlike heparin, requires no cofactor and is more effective in accessing clot-bound thrombin, promoting dissolution of mural thrombi and utility for acute coronary syndrome or deep vein thrombosis. However, hirudin has some undesirable properties. The irreversible 1:1 binding nature of hirudin to thrombin, carries with it the risk of severe bleeding in patients with reduced renal function necessary for clearing the peptide (Greinacher and Warkentin, Reference Greinacher and Warkentin2008). Moreover, early reports of low antigenicity (in light of being only 65 amino acids in length) proved premature and risk of IgG-mediated anaphylaxis is 0·16% in re-exposed patients. Hirudin orthologues from other species, like Hirudo verbana or Asiaticobdella fenestrata, while retaining the requisite thrombin-binding abilities, may reveal substantially different binding affinities or antigenicities that can mitigate against unwanted side-effects of current hirudin treatment regimes. HyPhy reveals evidence of positive selection on the molecule as a whole (P=0·0116) and that (excluding the signal peptide region) cysteines at positions 6 and 39, a glycine at position 10, and a phenylalanine at position 56 are under strong pressure not to change. Notably, the two cysteines are the first and last of six involved in forming the three disulphide bonds in the hirudin core, a region that also contains the constrained glycine and which inhibits the fibrinogen-binding exosite of thrombin. The constrained phenylalanine corresponds to that portion of the N-terminal domain of hirudin blocking the catalytic pocket of thrombin (Markwardt, Reference Markwardt1992). Bayesian network models reveal compensatory changes involving asparagine for non-adjacent residues known to associate with the Asp-His-Ser triad in the catalytic pocket of thrombin (Fig. 5).

Fig. 5. Evolutionary relationships (A) for single-copy transcripts of thrombin-inhibiting hirudin orthologues from hirudiniform leeches as determined by model-based analyses in HyPhy suggest that the protein is under strong positive selection (P<0·05). Hirudin, hirullin and haemadin had previously been characterized from European medicinal, Asian medicinal and Indian terrestrial leeches, respectively. Transcripts 3A0911, 4C08143 and 7B08185 were newly characterized from North American, European and African medicinal leeches, respectively. All divergences correspond to leech relationships depicted in Fig. 1. Fixed effects likelihood estimates indicate strong purifying selection (green) on two cysteines involved in disulfide bonds of the hirudin core, as well as on residues (G and F) associated with each of the two thrombin-binding domains of hirudin. Bayesian evolutionary networks revealed compensatory evolutionary changes in which an asparagine is required in exactly one of two amino acid positions that associate with thrombin's catalytic site (red).
This sort of comparative phylogenetic approach to understanding molecular function could prove more cost effective and more robust than alternatives like X-ray crystallography. The protease inhibiting antistasin family is revealing in this regard (Fig. 6). Antistasin has fully 20 cysteines involved in 10 disulphide bonds. Crystallography of antistasin complexed with factor Xa indicate that residues between the 17th and 18th cycteines in the C-terminal domain 2 of the protease inhibitor, are involved in the reactive site (Lapatto et al. Reference Lapatto, Krengel, Schreuder, Arkema, de Boer, Kalk, Hol, Grootenhuis, Mulders, Dijkema, Theunissen and Dijkstra1997). Likelihood ratio plots from FEL analysis pinpoint this same region as one with codons having an overall evolutionary history of strong negative selection (Fig. 6), but within which there is a residue adjacent to the 18th cysteine that is under strong positive (i.e. Darwinian) selection having changed nine times in the course of leech evolution. Taken together these results point to this region as being sufficiently significant in the history of the molecule's functioning that most of the amino acid residues are under strong pressure not to change. However, there may also be a single residue that may be responsible for the various changes in binding affinity; changes associated with switches from inhibiting factor Xa, to factor XIIIa, to elastase or to kallikrein.
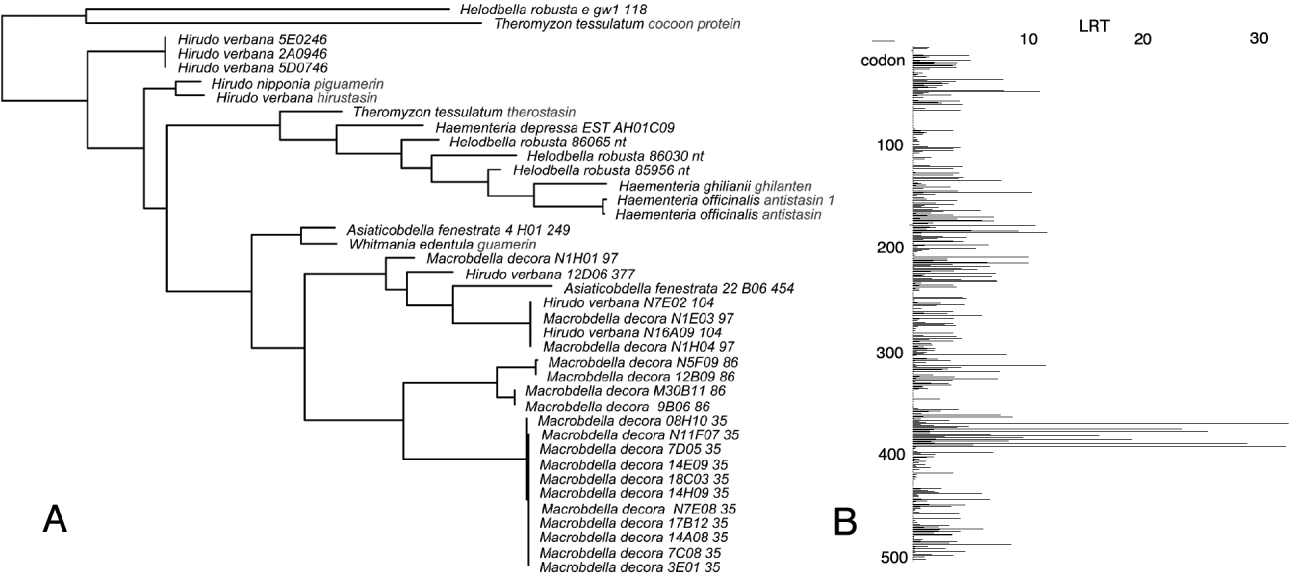
Fig. 6. Evolutionary relationships (A) for mutiple-copy transcripts of cysteine-rich antistasin family protease inhibitors from across leech diversity as determined by HyPhy and in which there is little overall concordance with leech phylogeny. A region exhibiting negative selection was detected (B) in the second antisasin domain in which likelihood ratios exceeded 10, and within which there was a single residue under Darwinian (positive) selection.
While 15 years of concerted effort has now provided a broad and compelling picture of the phylogenetic relationships of a wide array of parasitic groups from flatworms to roundworms and lice to leeches, it would be a shame if the utility of those analyses and the resulting trees were limited to mere systematic circumscription of natural groups with stable taxonomic names. While that was, perhaps, the initial raison d'etre of Phylogenetic Systematics, the toolbox available to phylogeneticists has become considerably more rich. With respect to the crop endosymbionts of medicinal leeches, it is the leech tree that adds depth and context to interpretation of the bacterial trees, and yet in neither case in a manner that corresponds to correlated co-speciation. In terms of potential biomedically relevant salivary secretions from leeches, it is again, in part, the leech phylogeny that adds a historically correlative framework for comparison; but also the emergent phylogeny of the proteins themselves. Together these trees within trees, and the power they provide for tracking and interpreting molecular change lead to enhanced understanding of evolutionary forces and ultimately function at the molecular level. We anticipate that these layers of intellectual pursuit driven by phylogenetic perspectives will only become richer, and the tools for their elucidation more intricate, as whole genomes of parasitic taxa (and their hosts) become more readily (and cheaply) available.
ACKNOWLEDGEMENTS
This research was supported by the US National Science Foundation, and by the Stavros Niarchos Foundation. We thank Sebastian Kvist and Alejandro Oceguera Figueroa for reviews of earlier drafts of portions of this contribution.






