


Introduction
Chagas disease, also known as American trypanosomiasis, is an important public health problem in Latin America and it is increasingly spreading in other areas. It is estimated that around six million people are affected worldwide and approximately 7000 deaths occur annually (WHO, 2016). The current treatment for Chagas disease includes two nitro-heterocyclic drugs – benznidazole (Bz) and nifurtimox – which are only effective against the parasite in the acute phase of infection. The use of these drugs is limited due to their poor bioavailability and side-effects, such as allergic dermatitis, pruritus and gastrointestinal manifestations among others (WHO, 2016; Chatelain, Reference Chatelain2017). These facts highlight an urgent need for the development of more effective and safer drugs to be used alone or in combination with other chemotherapeutic agents for the treatment of Chagas disease.
In the last decades, several antimicrobial peptides (AMPs) have been discovered. AMPs are naturally designed to target the cell membranes of pathogens and are widely seen as a promising alternative to conventional antimicrobials considering the rise in multi-antibiotic resistance (Sani and Separovic, Reference Sani and Separovic2016). Cathelicidins comprise one of these AMP families and are characterized by a conserved cathelin domain and a variable C-terminal cationic antimicrobial domain (Wódz and Brzezińiska-Błaszczyk, Reference Wódz and Brzezińiska-Błaszczyk2015). Recently, our research group have found and characterized cathelicidin precursors from the venom gland cDNA libraries of several species of South American pit viper snakes, called vipericidins (Rádis-Baptista, Reference Rádis-Baptista, Gopalakrishnakoneal, Inagaki, Mukherjee and Rahmy Carl-Wilhelm Vogel2015). Crotalicidin (Ctn), a vipericidin from the Crotalus durissus terrificus, has shown antibacterial (Falcão et al. Reference Falcao, de La Torre, Pérez-Peinado, Barron, Andreu and Rádis-Baptista2014) and antiproliferative activities against several cancer cell lines (Falcão et al. Reference Falcao, Pérez-Peinado, de la Torre, Mayol, Zamora-Carreras, Jiménez, Rádis-Baptista and Andreu2015). In silico dissection of Ctn yielded the fragments Ctn[1–14] and Ctn[15–34], which were tested to establish to what extent they reproduced the structure and activity of the parent peptide. Indeed, Ctn[15–34] displayed antibacterial, anti-tumour (Falcão et al. Reference Falcao, Pérez-Peinado, de la Torre, Mayol, Zamora-Carreras, Jiménez, Rádis-Baptista and Andreu2015) and antifungal activity (Cavalcante et al. Reference Cavalcante, Falcão, Fontenelle, Andreu and Rádis-Baptista2016) with very low haemolytic effect (Falcão et al. Reference Falcao, Pérez-Peinado, de la Torre, Mayol, Zamora-Carreras, Jiménez, Rádis-Baptista and Andreu2015). Recently, Ctn antiviral activity [15–34] was also demonstrated in an in vitro model of shrimp haemocytes (Vieira-Girão et al. Reference Vieira-Girão, Falcão, Rocha, Lucena, Costa and Rádis-Baptista2017).
The trypanosomicidal activity of animal venom components has been described by other authors (Adade et al. Reference Adade, Oliveira, Pais and Souto-Padrón2013; Souza et al. Reference Souza, Faria, Calabrese, Hardoim, Taniwaki, Alves and De Simone2016). In general, these venom antimicrobial components are amphipathic molecules mainly constituted of positively charged (hydrophilic) and hydrophobic residues, which can interact with and disrupt parasite cell membranes (Mcgwire and Kulkarni, Reference Mcgwire and Kulkarni2010; Teixeira et al. Reference Teixeira, Feio and Bastos2012). Thus, the aim of this work was to evaluate the antichagasic effect of cathelicidin Ctn and its short fragments (Ctn[1–14] and Ctn[15–34]) in comparison with the paradigmatic human cathelicidin LL-37 (hCAP18). Additionally, the main mechanisms of Ctn and Ctn peptides that trigger T. cruzi cell death were also investigated.
Materials and methods
Peptides and Bz
Ctn, Ctn[1–14] and Ctn[15–34] were obtained as described by Falcão et al. (Reference Falcao, de La Torre, Pérez-Peinado, Barron, Andreu and Rádis-Baptista2014, Reference Falcao, Pérez-Peinado, de la Torre, Mayol, Zamora-Carreras, Jiménez, Rádis-Baptista and Andreu2015). The human cathelicidin LL-37/hCAP18 was synthesized according to Falcão et al. (Reference Falcao, de La Torre, Pérez-Peinado, Barron, Andreu and Rádis-Baptista2014). The 1 mm peptide stock solutions were prepared by separately weighting and dissolving the right amount of each peptide in deionized water and kept at 4 °C for up to 6 weeks. Their amino acid sequences were analysed using the ‘Peptide property calculator’ (http://www.pepcalc.com) and ‘Heliquest’ (http://heliquest.ipmc.cnrs.fr) software programmes to obtain some physicochemical properties. Bz (C12H12N4O3; MW = 260.249 g mol−1) was obtained from Roche® (Basileia, Switzerland) and stock solutions were prepared with 1000 mm (1 M) of PBS and kept at 4 °C until its use within 6 weeks.
Trypanosoma cruzi and LLC-MK2 cell cultures
The Rhesus monkey kidney cells LLC-MK2 (ATCC CCL-7) were cultured in DMEM medium (Vitrocell, São Paulo, Brazil) supplemented with 10% fetal calf serum (FBS) and 1% antibiotics (100 U mL−1 penicillin and 100 µg mL−1 streptomycin) solution in T-25/75 cm2 flasks and were maintained at 37 °C in a humidified atmosphere with 5% CO2. These cells were subcultured each time they reached 80–90% of confluence after being harvested with a solution containing 0.25% of trypsin and 2.21 mm EDTA. Trypanosoma cruzi Y strain were isolated at the Chemistry Institute, Department of Biochemistry of the University of São Paulo.
Anti-T. cruzi activity assay against epimastigote forms
Epimastigote forms of T. cruzi Y strain were added to 96-well plates (106 parasites mL−1) containing 2-fold serial dilutions of Ctn, or Ctn[1–14], or Ctn[15–34], or LL-37 or Bz in liver infusion tryptose medium supplemented with 1% antibiotics (100 U mL−1 penicillin and 100 µg mL−1 streptomycin) and 10% of FBS. Final concentration ranges were: peptides at 0.19–100 µ m and Bz at 15.6–1000 µ m. Parasite growth inhibition was quantified in a Neubauer chamber after incubation at 28 °C for 24, 48 and 72 h (Rodrigues et al. Reference Rodrigues, Ueda-Nakamura, Corrêa, Sangi and Nakamura2014). Relative viability was determined with parasites treated only with sterile PBS medium as negative controls (100% viability) and all experiments were carried out in triplicate.
Anti-T. cruzi activity assay against trypomastigote forms
T. cruzi trypomastigote forms were obtained by infecting LLC-MK2 cells with trypomastigote forms in T-25/75 cm2 flasks at 37 °C in an atmosphere with 5% CO2 in DMEM medium (Vitrocell, São Paulo, Brazil) supplemented with 1% antibiotics (100 U mL−1 penicillin and 100 µg mL−1 streptomycin) and 2% FBS for 24 h. Thereafter, trypomastigote forms were plated (96-well plate) and 2-fold serial dilutions of either peptides or Bz in DMEM were added at the same final concentration ranges used against the epimastigote forms. After 24 h of incubation at 37 °C, parasite growth inhibition was quantified in a Neubauer chamber (Adade et al. Reference Adade, Carvalho, Tomaz, Costa, Godinho, Melo, Lima, Rodrigues, Zingali and Souto-Padrón2014). Relative cell viability was calculated with cells treated only with sterile PBS medium as negative controls and all experiments were carried out in triplicate.
Anti-T. cruzi activity assay against amastigote forms
LLC-MK2 cells were seeded in 24-well plates (5 × 105 cells/well) containing glass coverslips (13-mm diameter) in DMEM medium supplemented with 10% FBS and 1% antibiotics (100 U mL−1 penicillin and 100 μg mL streptomycin), and maintained for 24 h at 37 °C in a 5% CO2 atmosphere. Next, the medium was removed from the wells and LLC-MK2 cells were infected with trypomastigote forms (parasite–host cell ratio of 20:1) in DMEM medium containing 2% FBS. After 48 h of incubation, the cells were washed to remove the non-internalized parasites and treated with either Ctn (0.22 or 0.44 µ m) or Bz (282 or 564 µ m) in DMEM medium with 2% FBS. After incubation for 24 or 48 h at 37 °C, the coverslips were collected, washed with PBS, fixed in Bouin's solution and stained with Giemsa (Lima et al. Reference Lima, Sousa, Torres, Rodrigues, Mello, Menezes, Tessarolo, Quinet, de Oliveira and Martins2016). Non-treated infected LLC-MK2 cells were used as controls. 300 cells were counted in triplicate and intracellular amastigotes per 100 cells and percentage of infected cells were calculated.
Trypanosoma cruzi epimastigote death pathway assays
Flow cytometry experiments were performed to evaluate T. cruzi death pathways when treated with Ctn. The epimastigote forms (106 cells mL−1) were treated with Ctn and Bz at their half maximal effective concentration (EC50) concentrations (4.47 and 218 µ m, respectively) in liver infusion tryptose medium. After incubation for 24 h at 28 °C, the parasite cells were washed and stained with fluorescein isothiocyanate-annexin V (AX) and/or 7-aminoactinomycin D (7-AAD) according to the manufacturer's instructions (BD Pharmingen, Franklin Lakes, NJ, USA) to detect cell death by apoptosis or necrosis, respectively. In separate experiments, the loss of mitochondrial transmembrane potential (MTP), the outburst of reactive oxygen species (ROS) and the swelling of reservosomes were also investigated. After the treatment of T. cruzi epimastigote forms with Ctn and Bz at their EC50 (4.47 and 218 µ m, respectively) for 24 h at 28 °C, the parasites were stained with 10 µg mL−1 rhodamine 123 or 20 mm L−1 dichloro-dihydro-fluorescein diacetate (DCFH-DA) or 5 µg mL−1 acridine orange (AO) (Sigma – Aldrich™, St. Louis, MO, USA), according to the manufacturer's instructions, respectively. Next, these cells were run in a FACSCalibur flow cytometer and 10 000 live events were analysed using the Cell Quest software (Becton-Dickinson, San Jose, CA, USA). The results were established by determining the fold change (treated/non-treated cell ratio) of the geometric mean of fluorescence. Epimastigotes without Ctn treatment were used as controls and all experiments were carried out in triplicate.
Cytotoxicity test in mammalian cells (MTT assay)
Cell viability was also measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Vanden Berghe et al. Reference Vanden Berghe, Grootjans, Goossens, Dondelinger, Krysko, Takahashi and Vandenabeele2013). LLC-MK2 cells were plated in DMEM medium, treated with different concentrations of Ctn, Ctn[1–14] and Ctn [15–34] (1.56–100 µ m) and incubated at 37 °C for 24 h. MTT (Amresco, Ohio, USA; 5 mg mL−1) was added and the cells were incubated for 4 h, when 10% sodium dodecyl sulphate (Vetec, São Paulo, Brazil) was added to solubilize the formazan product. Cell viability measurements were performed at 570 nm on a microplate reader (Biochrom® Asys Expert Plus). Relative cell viability was calculated using cells treated only with sterile PBS medium as negative controls and all experiments were carried out in triplicate. The Selectivity Index (SI) was calculated through the ratio of EC50 of LLC-MK2 cells/EC50 of trypomastigote forms (Nwaka and Hudson, Reference Nwaka and Hudson2006).
Scanning electron microscopy
Epimastigote forms were treated with Ctn peptide at its EC50 (4.47 µ m) concentration in liver infusion tryptose medium. After incubation for 12 h at 28 °C, the parasites were washed with PBS and fixed with a 2.5% glutaraldehyde (Electron Microscopy Sciences, Hatfield, PA, USA) solution. After fixation for 2 h at room temperature, parasites were washed with PBS, and then dehydrated with ethanol. Next, samples were dried with CO2, coated in gold and, finally, observed in a FEI Quanta 450 FEG scanning electron microscope (FEI, Oregon, USA). Digital images were acquired and stored (Lima et al. Reference Lima, Sousa, Torres, Rodrigues, Mello, Menezes, Tessarolo, Quinet, de Oliveira and Martins2016).
Statistical analysis
Three independent experiments were performed in triplicate for all assays. The statistical analysis was performed using the GraphPad Prism 5 programme (GraphPad Software, San Diego, CA, USA). The EC50 values were determined by non-linear regression. Data were analysed using one-way analysis of variance (ANOVA) followed by Dunnett's post-test. Significance was defined as *P < 0.05.
Results
The obtained peptides with their respective amino acid sequences and physicochemical properties such as net charge, hydrophobicity and molecular weight are shown in Table 1. They were screened against all T. cruzi morphological forms to assess and compare their antiprotozoa activities with the standard drug Bz.
Table 1. Amino acid sequence and some physicochemical properties of the peptides

a Ctn and fragments had C-terminal amides.
b At neutral pH, from the ‘Peptide property calculator’ software (http://www.pepcalc.com).
c Obtained from the ‘Heliquest’ software (http://heliquest.ipmc.cnrs.fr).
d Previously determined (Falcão et al. Reference Falcao, Pérez-Peinado, de la Torre, Mayol, Zamora-Carreras, Jiménez, Rádis-Baptista and Andreu2015).
Against the epimastigote forms, the only peptide that showed activity against T. cruzi within the tested concentration range was Ctn, with an IC50 of 4.47 (±0.9) μM reached after 24 h of incubation, which did not change up to 72 h after treatment. Bz required very high molar concentrations, as expected, against the Y strain (IC50/24 h of 218 ± 15 µ m) (Table 2) and could not reach an IC50 value close to Ctn concentrations even after 72 h of treatment (IC50/72 h of 16.5 ± 1 µ m).
Table 2. EC50 values (in μ m) and Selectivity Index (SI) to trypomastigote forms of the peptides and Bz against epimastigote and trypomastigote forms of Trypanosoma cruzi Y strain and LLC-MK2 mammalian cells
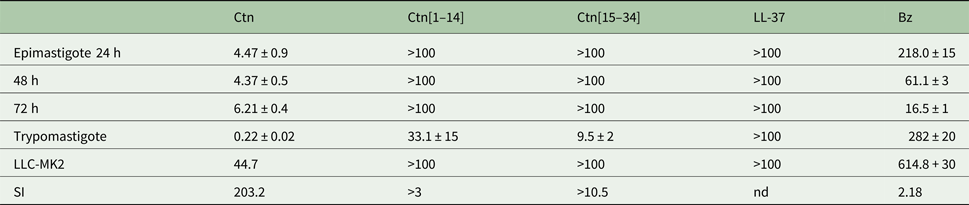
Data are expressed as means ± s.e.m. of triplicate of three independent experiments (nd: not determined).
Against the trypomastigote forms, Ctn and its fragments showed different activities against T. cruzi. While Ctn[1–14] and Ctn[15–34] was able to inhibit 50% of the trypomastigotes with concentrations of 33.1 and 9.5 µ m, respectively, Ctn only required 0.22 µ m to inhibit 50% of that T. cruzi morphological form. Next, the cytotoxicity of the peptides to LLC-MK2 cells without the parasites was verified.
T. cruzi amastigote forms inside LLC-MK2 were also treated with Ctn or Bz at their respective EC50s against the trypomastigote forms. According to Fig. 1, Ctn significantly reduced the number of amastigotes for each 100 LLC-MK2 cells when compared to untreated infected cells (control) after 24 h of incubation (Fig. 1A). In addition, Ctn at 0.22 µ m significantly reduced the percentage of infected LLC-MK2 cells when compared to the control after 48 h of treatment (Fig. 1B). Similar results were also observed with Bz at 282 µ m.
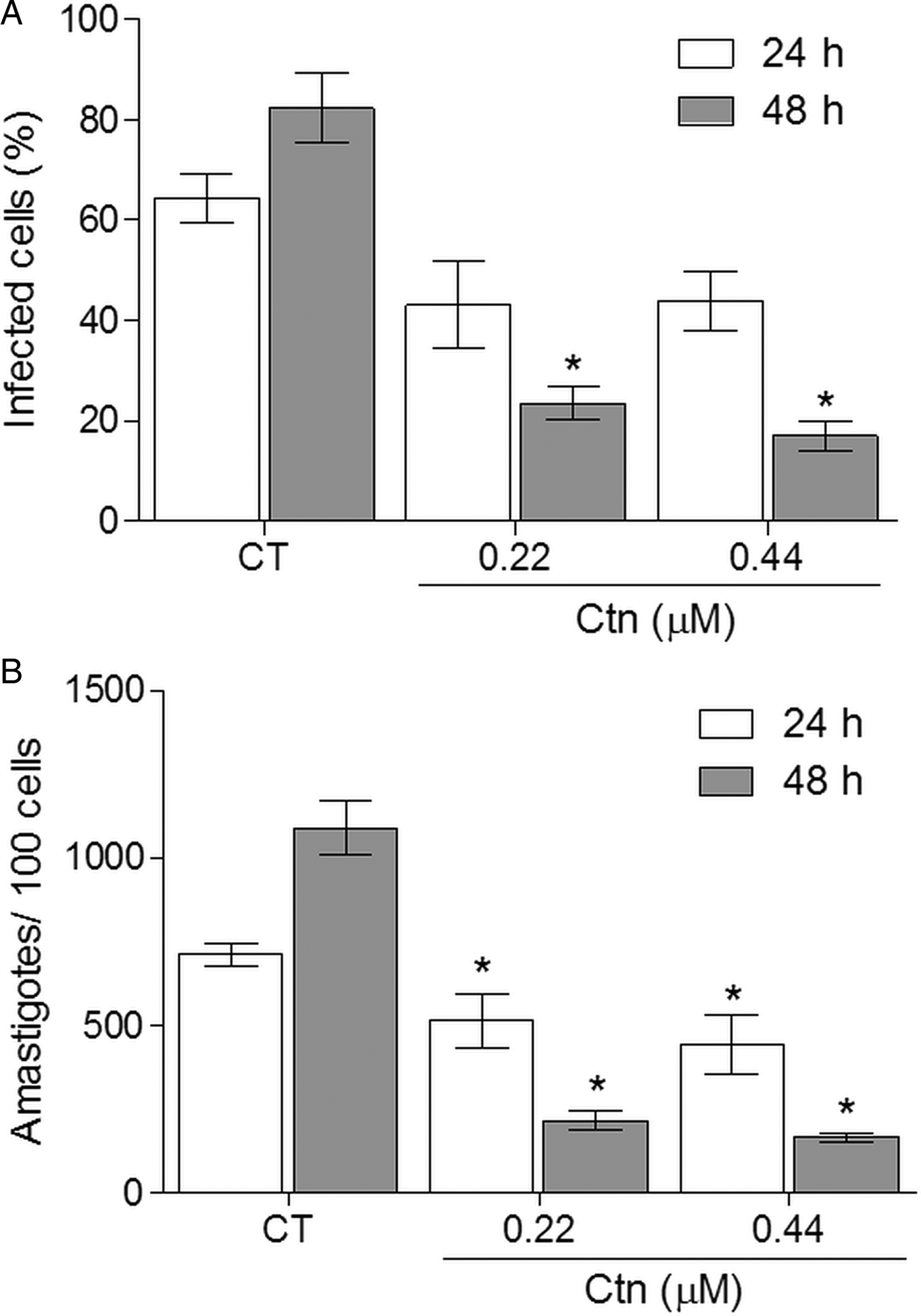
Fig. 1. Effect of crotalicidin on intracellular amastigotes in infected cells (A) amastigote/100 cells (B) at 24 and 48 h of incubation (CT – control treated with PBS; *P < 0.05).
In addition, flow cytometry analyses and scanning electron microscopy (SEM) were performed to evaluate the death pathway(s) of T. cruzi epimastigote forms. After 24 h of treatment with Ctn at 4.4 µ m (EC50), the remaining live epimastigotes were mainly labelled with either 7-AAD only (41.6%) or 7-AAD and AX (39.6%) markers, which indicate cell death by necrosis or necrosis/late apoptosis, respectively (Fig. 2). Bz was able to increase the labelling with 7-AAD and AX (8.0%) and more significantly the labelling with 7-AAD only (65.6%) on epimastigotes treated with the EC50 after 24 h (Supplementary Fig. S1A).
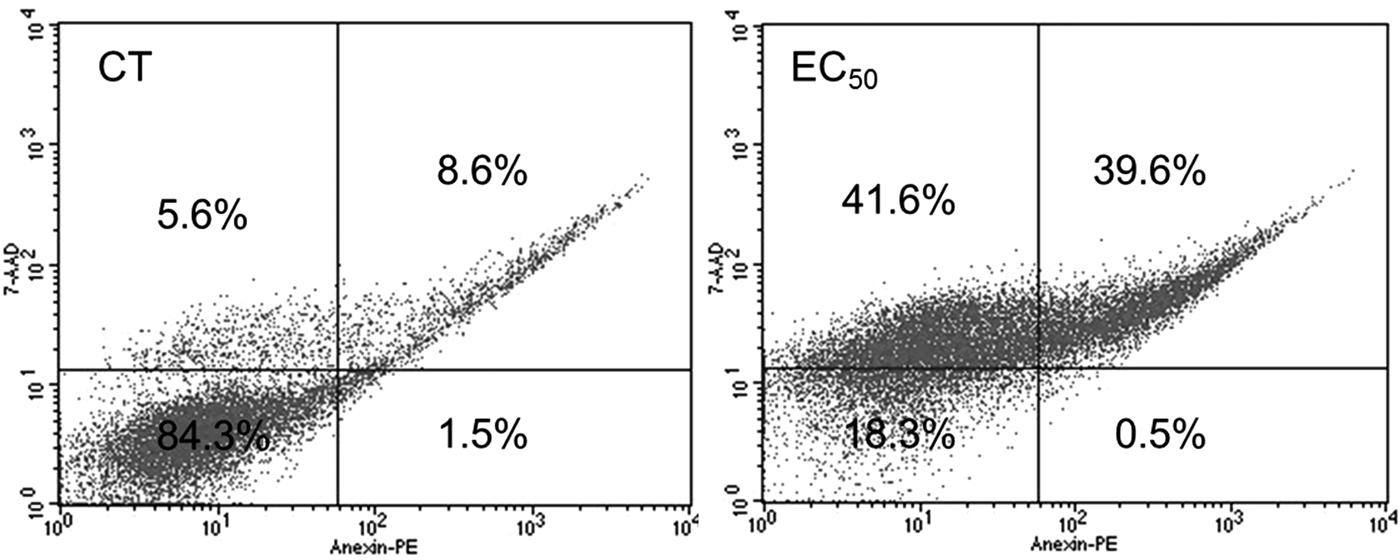
Fig. 2. Representative flow cytometry scattering graphs of Trypanosoma cruzi epimastigotes treated with crotalicidin at 4.47 µ m for 24 h. Percentage values are averages of 10 000 live events from three independent experiments (CT – control treated with PBS).
Additional flow cytometry experiments (Fig. 3) were then carried out to clarify the epimastigote death pathway, such as the generation of ROS and the loss of MTP. In Fig. 3A, Ctn at 4.47 µ m for 24 h induced a burst of ROS, increased DCFH-DA signal (shift to the right), by as much as 3-fold when compared to untreated parasites (control). In a separate assay, a reduction in MTP was observed, decreasing rhodamine 123 signal by 50% (shift to the left), when the epimastigotes were treated with Ctn at the EC50 compared to non-treated T. cruzi (Fig. 3B). Bz showed a similar profile, decreasing MTP by approximately 30% and increasing by 70% ROS generation in epimastigotes treated with the EC50 after 24 h (Supplementary Fig. S1B–C).

Fig. 3. A representative histogram of DCFH-DA fluorescent signal and relative fluorescence values (from three independent experiments) from Trypanosoma cruzi epimastigotes that were initially treated with crotalicidin at 4.47 µ m for 24 h (A) and a representative histogram of rhodamine 123 (Rho 123) fluorescent signal and relative fluorescence values (from three independent experiments) from T. cruzi epimastigotes initially treated with crotalicidin at 4.47 µ m for 24 h (B) (CT – control treated with PBS).
We also investigated an alternative cell death pathway, the autophagy, utilizing AO staining to evaluate the increase of acidic vesicles, the reservosomes. However, after 24 h, epimastigote forms treated with Ctn at the EC50 did not show any increase in the AO fluorescent signal compared to the control group (Supplementary Fig. S2), neither did Bz (Supplementary Fig. S1D).
Based on the abovementioned results, which provided an insight that Ctn kills T. cruzi by inducing necrosis and late apoptosis, SEM images (Fig. 4) were acquired to visualize the morphological alterations and, thus corroborate the flow cytometry findings. Compared to a non-treated T. cruzi epimastigote (Fig. 4A), parasites treated for 12 h with Ctn at the EC50 showed cell shrinkage (Fig. 4B) and loss of membrane integrity with the presence of pores (Fig. 4C–D).

Fig. 4. Scanning electron microscopy of control epimastigotes (A) and treated epimastigotes with EC50 of crotalicidin (B–D). Morphological alterations were observed, such as cell shrinkage (B) and membrane integrity loss (C–D). Scale bars: 1 µm.
Discussion
In this present study, we assessed the potential anti-T. cruzi activity of full-size Ctn, Ctn[1–14] and Ctn[15–34] short Ctn derivatives and LL-37 against all parasite morphological forms and compared them with Bz, a commercially available antichagasic drug of choice, as well as studied the death pathway(s) induced by the most promising biologically active peptide. Against the epimastigotes, Ctn was the only peptide that showed activity at the tested concentration range. By observing the physicochemical properties of the analysed peptides, it seems that both bulky and highly positively charged peptides are required for anti-T. cruzi activity against the epimastigote forms. Similar results against T. cruzi were previously reported with the use of batroxicidin (BatxC), a vipericidin from Bothrops atrox that has a molecular weight of 4258.63 and net charge of +16 (Falcao et al. Reference Falcao, de La Torre, Pérez-Peinado, Barron, Andreu and Rádis-Baptista2014; Mello et al. Reference Mello, Lima, Menezes, Bandeira, Tessarolo, Sampaio, Falcão, Rádis-Baptista and Martins2017). In addition, after 24 h of treatment, Ctn EC50 (4.47 µ m) was more than one order of magnitude lower than Bz IC50 (218 µ m), which did not reach values close to the peptide even after 72 h of incubation.
The peptides were also evaluated against trypomastigotes and amastigotes. These T. cruzi morphological forms were obtained after infecting LLC-MK2 kidney cells, which have been used for more than 30 years as mammalian cell models of T. cruzi infection (Andrews and Colli, Reference Andrews and Colli1982; Zingales et al. Reference Zingales, Andrews, Kuwajima and Colli1982; Zingales et al. Reference Zingales, Pereira, Almeida, Umezawa, Nehme, Oliveira, Macedo and Souto1997). Interestingly, while the in vitro Bz concentration killed 50% of the trypomastigotes (282 µ m), it essentially did not change when compared to epimastigotes after 24 h of incubation. In contrast, the Ctn EC50 against the trypomastigote forms (0.22 µ m) showed a 20-fold lower effect against epimastigotes (4.47 µ m). Moreover, Ctn at 0.22 µ m also significantly reduced the number of amastigotes and the percentage of infected LLC-MK2 cells when compared to non-treated infected cells. Therefore, T. cruzi trypomastigote and amastigote forms seem to be more susceptible to Ctn than epimastigotes. This increased susceptibility can be related to the prevalence of negatively charged molecules on the parasite cell membrane, especially in trypomastigote forms, such as mucin glycoproteins and sialic acid, which are implicated in host–parasite interactions (de Souza et al. Reference De Souza, De Carvalho and Barrias2010). These anionic molecules made T. cruzi trypomastigote forms susceptible even to Ctn[1–14] and Ctn[15–34] fragments, which have only half of the net charge of Ctn, particularly the mid-sized and highly hydrophobic Ctn[15–34].
The cytotoxicity of the drugs (peptides and Bz) against uninfected LLC-MK2 cells was also verified. Considering the IC50 concentrations against trypomastigotes, Ctn showed the greatest selectivity (SI = 203.2) among the other peptides and Bz. Furthermore, Ctn met two important criteria for a potential anti-T. cruzi drug candidate to be considered as a success and, thus, the most promising evaluated peptide: trypomastigote IC50 below 1 µg mL−1 (0.22 µ m or 0.913 µg mL−1) and SI above 50 (Nwaka and Hudson, Reference Nwaka and Hudson2006). Furthermore, the Ctn selectivity against parasite-infected cells seems to be another advantage for the clinical development of target therapy for T. cruzi infection.
The necrotic pathway observed in Ctn was also observed when the epimastigotes were treated with the vipericidin BatxC at the EC50 (Mello et al. Reference Mello, Lima, Menezes, Bandeira, Tessarolo, Sampaio, Falcão, Rádis-Baptista and Martins2017). Trypanosoma cruzi cell death by apoptosis and autophagy was also observed when other AMPs were used, such as melittin (Adade et al. Reference Adade, Oliveira, Pais and Souto-Padrón2013).
Cathelicidins are typically potent trypanolytic AMPs. Similar peptides such as cathelicidins SMAP-29 and protegrin-1 have been tested in the intraperitoneal route and showed interesting results, such as decrease in parasitaemia and prolonged survival of mice infected with Trypanosoma brucei (McGwire et al. Reference McGwire, Olson, Tack and Engman2003). The evaluation of Bz-induced cell death in epimastigote forms also showed a necrotic profile, meaning Ctn is promising as a template for the designing of a new antichagasic agent, because it has a similar cell death profile to the reference drug and an almost 10-fold higher selective index.
In conclusion, Ctn, a cathelicidin-related peptide obtained from the venom gland of C. durissus terrificus rattlesnake, not only has antibacterial and anti-tumour properties but also exhibited anti-T. cruzi activity against all morphological forms of a Bz-resistant strain. In addition, Ctn killed the epimastigote parasite form by inducing necrosis. Remarkably, when toxicity to healthy mammalian cells was compared, the peptide's greater selectivity for T. cruzi when compared to Bz makes Ctn a promising drug candidate or a lead for the development of new peptide-based drugs to treat Chagas disease.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182017001846
Acknowledgements
C.B. Falcão is an associate researcher from the National Program for Post-Doctorates (CAPES/ME) at the Postgraduate Program in Pharmaceutical Sciences, Faculty of Pharmacy, Dentistry and Nursing, the Federal University of Ceará. The authors also thank Analytic Centre – UFC/CT – INFRA/Pro – Equipamento CAPES.
Financial support
This research study was partially supported by the Toxinology Program (Issue 2010), the Coordination for the Improvement of Higher Education Personnel (CAPES), the Ministry of Education and Culture (MEC) of the Federal Government of Brazil, granted to Prof G. Rádis-Baptista.








