Introduction
Tsangpoite, Ca5(PO4)2(SiO4) (P63 or P63/m; a = 9.489(4) Å, c = 6.991(6) Å, V = 545.1(6) Å3 and Z = 2), the hexagonal polymorph of silicocarnotite, and matyhite, Ca9(Ca0.5□0.5)Fe(PO4)7 (R3c, a = 10.456(7) Å, c = 37.408(34) Å, V = 3541.6(4.8) Å3 and Z = 6), the Fe-analogue of Ca-merrillite, were identified from the D'Orbigny angrite meteorite in this study. Angrites are a small group of achondritic meteorites of basaltic composition, and are generally believed to be formed by rapid crystallisation of unusual Ca-, Al-, Ti-rich and Na-, K-poor magmas at the very early stage of solar system history (Prinz et al., Reference Prinz, Keil, Hlava, Berkley, Gomes and Curvello1977; Goodrich, Reference Goodrich1988; McKay et al., Reference McKay, Lindstrom, Yang and Wagstaff1988, Reference McKay, Crozaz, Wagstaff, Yang and Lundberg1990; Mittlefehldt and Lindstrom, Reference Mittlefehldt and Lindstrom1990; Prinz and Weisberg, Reference Prinz and Weisberg1995; Mikouchi et al., Reference Mikouchi, Miyamoto and McKay1996; Mikouchi and McKay, Reference Mikouchi and McKay2001; Mittlefehldt et al., Reference Mittlefehldt, Killgore and Lee2002; Keil, Reference Keil2012). Angrites consist mainly of anorthite, olivine, Al–Ti augite and kirschsteinite, as well as grain-boundary phases such as Al–Ti-rich hedenbergite, Ca–Fe-rich olivine, Fe sulfide (FeS, troilite), ulvöspinel, and unusual minerals such as kuratite (the Fe-analogue of rhönite, IMA2013-109; described as Ti-silicate or rhönite in Mittlefehldt et al., Reference Mittlefehldt, Killgore and Lee2002; Kurat et al., Reference Kurat, Varela, Brandstätter, Weckwerth, Clayton, Weber, Schultz, Wäsch and Nazarov2004; Jambon and Boudouma, Reference Jambon and Boudouma2011; Hwang et al., Reference Hwang, Shen, Chu, Yui, Varela and Iizuka2016a), and some silico-phosphates with variable SiO2, characteristically high FeO, and very low F and Cl contents (see table 8 in Keil, Reference Keil2012).
Tsangpoite, referred to as a Ca silico-phosphate with ~10–15 wt.% SiO2 and ~25–35wt.% P2O5 in Kaneda et al. (Reference Kaneda, Mikouchi, Saito, Sugiyama, Ohsumi, Mukai, Osaka, Miyata, Nakai, Kasama, Chikami and Miyamoto2001), was first reported in Asuka 881371 (Prinz and Weisberg, Reference Prinz and Weisberg1995; Warren and Davis, Reference Warren and Davis1995), and later in other samples such as D'Orbigny (Kaneda et al., Reference Kaneda, Mikouchi, Saito, Sugiyama, Ohsumi, Mukai, Osaka, Miyata, Nakai, Kasama, Chikami and Miyamoto2001; Mittlefehldt et al., Reference Mittlefehldt, Killgore and Lee2002), NWA 1296 & 1670 (Jambon et al., Reference Jambon, Barrat, Boudouma, Fonteilles, Badia, Göpel and Bohn2005; 2008) and NWA 4590 (Mikouchi et al., Reference Mikouchi, Sugiyama, Satake and Amelin2011). However, due to the low abundance and small crystal size, as well as variable SiO2 and FeO contents, determination of the stoichiometry of tsangpoite in most cases was inconclusive. It was noted that the compositions of tsangpoite are close to silicocarnotite or nagelschmidtite, but slightly different from both of them (e.g. Kaneda et al., Reference Kaneda, Mikouchi, Saito, Sugiyama, Ohsumi, Mukai, Osaka, Miyata, Nakai, Kasama, Chikami and Miyamoto2001; Mikouchi et al., Reference Mikouchi, Sugiyama, Kato, Yamaguchi, Koizumi and Kaneda2010). Structurally, the early synchrotron X-ray Laue pattern of tsangpoite from Asuka 881371 did not match that of merrillite, apatite, silicocarnotite or ‘fassaite’ (Kaneda et al., Reference Kaneda, Mikouchi, Saito, Sugiyama, Ohsumi, Mukai, Osaka, Miyata, Nakai, Kasama, Chikami and Miyamoto2001). Micro-Raman and electron back-scatter diffraction (EBSD) analyses using scanning electron microscopy (SEM), however, showed that the tsangpoite from D'Orbigny could be isostructural to apatite (Mikouchi et al., Reference Mikouchi, Sugiyama, Kato, Yamaguchi, Koizumi and Kaneda2010). This is further supported by X-ray diffraction (XRD) of the exceptionally large tsangpoite crystals (a few hundreds of μm) with a high Fe3+/ΣFe ratio (~0.8; X-ray absorption near edge structure (XANES) analysis) showing an apatite-like unit cell and space group (Mikouchi et al., Reference Mikouchi, Sugiyama, Satake and Amelin2011). Further, in this present investigation we show that tsangpoite, with a hexagonal structure, a silicocarnotite-like stoichiometry, and abundant structure vacancies due to aliovalent cation substitution, is most probably structurally related to the α-Ca2SiO4 high-T polymorph, as are flamite, nagelschmidtite and silicocarnotite (Saalfeld and Klaska,Reference Saalfeld and Klaska1981; Gfeller et al., Reference Gfeller, Widmer, Krüger, Galuskin and Armbruster2015; Widmer et al., Reference Widmer, Gfeller and Armbruster2015; Galuskin et al., Reference Galuskin, Galuskina, Gfeller, Krüger, Kusz, Vapnik, Dulski and Dzierżanowski2016). This indicates a mineralogical origin for tsangpoite at T > 1200°C, similar to the high-temperature hexagonal phase(s) in the α-Ca2SiO4–![]() $\bar {\rm \alpha} $-Ca3(PO4)2 binary system, although an apatite-like structure with fully empty anion channels for tsangpoite cannot be excluded completely pending future single-crystal refinements. With the stoichiometry and structure unknown in other mineral species, tsangpoite should be considered as the first new mineral in the silicate–phosphate category in meteorites (Hwang et al., Reference Hwang, Shen, Chu, Yui, Varela and Iizuka2015; Rubin and Ma, Reference Rubin and Ma2017).
$\bar {\rm \alpha} $-Ca3(PO4)2 binary system, although an apatite-like structure with fully empty anion channels for tsangpoite cannot be excluded completely pending future single-crystal refinements. With the stoichiometry and structure unknown in other mineral species, tsangpoite should be considered as the first new mineral in the silicate–phosphate category in meteorites (Hwang et al., Reference Hwang, Shen, Chu, Yui, Varela and Iizuka2015; Rubin and Ma, Reference Rubin and Ma2017).
Matyhite, referred to as a Si-bearing Ca-phosphate (1–7 wt.% SiO2 and 35–45 wt.% P2O5) in Kaneda et al. (Reference Kaneda, Mikouchi, Saito, Sugiyama, Ohsumi, Mukai, Osaka, Miyata, Nakai, Kasama, Chikami and Miyamoto2001), was reported in plutonic angrites such as Angra dos Reis (Prinz et al., Reference Prinz, Keil, Hlava, Berkley, Gomes and Curvello1977), LEW 86010 (Mckay et al., Reference McKay, Lindstrom, Yang and Wagstaff1988), Asuka 881371 (Prinz and Weisberg, Reference Prinz and Weisberg1995; Warren and Davis, Reference Warren and Davis1995), D'Orbigny (Kaneda et al., Reference Kaneda, Mikouchi, Saito, Sugiyama, Ohsumi, Mukai, Osaka, Miyata, Nakai, Kasama, Chikami and Miyamoto2001; Mittlefehldt et al., Reference Mittlefehldt, Killgore and Lee2002) and NWA 1296 (Jambon et al., Reference Jambon, Barrat, Boudouma, Fonteilles, Badia, Göpel and Bohn2005). In contrast to tsangpoite, matyhite received less attention in the past, probably because it has long been recognised to have a merrillite-like stoichiometry, and its structure determination by the synchrotron Laue method was inconclusive (e.g. Kaneda et al., Reference Kaneda, Mikouchi, Saito, Sugiyama, Ohsumi, Mukai, Osaka, Miyata, Nakai, Kasama, Chikami and Miyamoto2001; Mikouchi et al. Reference Mikouchi, Kaneda, Miyamoto, Sugiyama and Ohsumi2001). In fact, as reported here, matyhite has a merrillite structure with high Ca, Fe, and low Na, Mg contents, in marked contrast to other known merrillite-group minerals from Martian meteorites or Lunar rocks (e.g. Britvin et al., Reference Britvin, Krivovichev and Armbruster2016; Hughes et al., Reference Hughes, Jolliff and Gunter2006, Reference Hughes, Jolliff and Rakovan2008; Jolliff et al., Reference Jolliff, Hughes, Freeman and Zeigler2006), and hence should be considered as a new merrillite-group mineral (Hwang et al., Reference Hwang, Shen, Chu, Yui, Varela and Iizuka2016b).
Tsangpoite is named in honour of Professor Dr. Tsang-Po Yen (1914–1994), former senior geologist of the Geological Survey of Taiwan (1946–1974) and director of the Institute of Geophysics, National Central University, Taiwan (1974–1981). Prof. Yen contributed immensely to the mineralogical, petrological, ore-deposit and tectonic studies in Taiwan, including mineral characterisation for igneous and metamorphic rocks, structural and stress analyses for metamorphic rocks, identifying high-pressure rocks and metamorphic-belt subdivision, as well as metal and non-metal ore explorations. He published more than 90 papers during his academic career, and was highly respected by the Earth science community in Taiwan.
Matyhite is named in honour of Professor Dr. Ting-Ying H. Ma (1899–1979, middle name Hsüeh (H.) meaning snowy peak in Chinese), a distinguished palaeontologist who pioneered research into relations between coral growth rate, sea-water temperature, paleoclimate and paleogeography, with major contributions during 1930–1960 (cf. Yang and Oldroyd, Reference Yang and Oldroyd2003). He was one of the early advocates of continental drift. After World War II, Prof. Ma went to Taiwan, and was appointed the joint Director of the Department of Geology and the Institute of Oceanography of National Taiwan University and served from 1946 to 1950.
Type materials of tsangpoite and matyhite (as well as kuratite) are deposited in the collections of the Naturhistorisches Museum Wien, Vienna, Austria, inventory number Section D'Orbigny C-N1172-NH Wien, and the National Museum of Natural Science, Taiwan, ROC, inventory number NMNS007600-P020440.
Samples and analytical methods
Two polished slab samples with inventory number: Section D´Orbigny C-N1172-NH Wien (~1.5 cm × 1.5 cm) and M1173 (~2.0 cm × 3.0 cm in size) from the porous part and the dense part of D'Orbigny angrite, respectively, were loaned by the Naturhistorisches Museum Wien, Austria for the present study. Petrography of the D'Orbigny angrite has been described in detail by Mittlefehldt et al. (Reference Mittlefehldt, Killgore and Lee2002), Varela et al. (Reference Varela, Kurat, Zinner, Métrich, Brandstätter, Ntaflos and Sylvester2003) and Kurat et al. (Reference Kurat, Varela, Brandstätter, Weckwerth, Clayton, Weber, Schultz, Wäsch and Nazarov2004).
Thin sections of the slab samples were studied by optical microscopy (OM) and SEM under back-scattered electron (BSE) mode coupled with energy-dispersive X-ray (EDX) analyses using a JEOL JSM-7000F instrument operated at 15 kV. Transmission electron microscopic (TEM) imaging, single-crystal electron diffraction and TEM-EDX analyses were carried out using a JEOL 3010 analytical electron microscope (AEM) operated at 300 kV. More than three dozen TEM thin sections for imaging and electron diffraction studies were prepared using a SEIKO SMI-3050 focus ion beam microscope. Selected area electron diffraction (SAED) patterns were obtained from μm-sized tsangpoite and matyhite crystals at 120 cm camera length. For each diffraction pattern, only two or three of the shortest nonlinear reciprocal vectors were measured for d-spacing calculations. In general, the largest d spacing among a row of reflections with multiple hkl data was used for unit-cell refinement. The error of the d-spacing measurements of SAED patterns taken at a camera length of 120 cm and calibrated with an Al standard was estimated to be ±0.002 nm. Unit-cell parameters were refined using least-squares from measured d spacings extracted from electron-diffraction patterns.
Chemical analyses were undertaken using a JEOL JXA8500-F FE electron microprobe using wavelength dispersive spectroscopy (WDS) mode at 12 kV and 5 nA with a 2 µm beam diameter. Natural and synthetic mineral standards and diffracting crystals (in parentheses) used for calibration were: diopside for SiKα (TAP); rutile for TiKα (PET); corundum for AlKα (TAP), chromium oxide for CrKα (PET); hematite for FeKα (LiF); tephroite for MnKα (PET); periclase for MgKα (TAP); Ni oxide for NiKα (LiF); wollastonite for CaKα (PET); albite for NaKα (TAP); adularia for KKα (PET); fluorapatite for PKα (PET); fluorite for FKα (TAP); tugtupite for ClKα (PET); celestine for SKα (PET); Zn oxide for ZnKα (LiF); celestine for SrLα (PET); Y-Al garnet (YAG) for YLα (PET); LaP5O14 for LaLα (LiF); CeP5O14 for CeLα (LiF); PrP5O14 for PrLβ (LiF); NdP5O14 for NdLβ (LiF); and YbP5O14 for YbLα (LiF). Counting times for the peak of each element and backgrounds both above and below the peak were 20 s and 10 s, respectively. The detection limit for trace elements using the K or L line is ~600 ppm or 2000 ppm, respectively. Potential peak overlapping between elements was filtered by appropriate selection of the baseline counting position by pulse height analyser on X-ray counter. Targets for electron-probe analysis were selected carefully by secondary and back-scattered electron images.
The Raman spectrum of minute tsangpoite and matyhite grains ~5–10 µm in size were conducted by using a LABRAM HR micro-Raman spectrometer equipped with an Ar+ laser with 514.5 nm excitation and a spatial resolution of 2–5 µm.
Results
General description of the D'Orbigny angrite and its phase assemblage
The texture of D'Orbigny can be described as a fluffy, but interlocking framework of anorthite (An) + olivine (Ol) (Fe-rich forsterite core and Ca-rich fayalite mantle) intergrowth plates of ~0.5–2 mm in thickness and up to ~5–10 mm in width/length. They are overgrown by a kirschsteinite (Kir) layer and/or a Ca-rich fayalite (Fa)–Ca-deficient kirschsteinite intergrowth layer, with interstitial space filled by augite (Aug) with an Al–Ti-bearing hedenbergite (Hd) rim as well as a grain-boundary phase assemblage (cf. SEM montage in Supplementary material: Fig. S1a,b). The grain-boundary phase assemblage consists of: Fe sulfide (FeS, troilite); ulvöspinel; kuratite (Ku) (Hwang et al., Reference Hwang, Shen, Chu, Yui, Varela and Iizuka2016a), described as Ti silicate or rhönite in the literature (Mittlefehldt et al., Reference Mittlefehldt, Killgore and Lee2002; Kurat et al., Reference Kurat, Varela, Brandstätter, Weckwerth, Clayton, Weber, Schultz, Wäsch and Nazarov2004; Jambon and Boudouma, Reference Jambon and Boudouma2011); new mineral tsangpoite (Tsa) (Hwang et al., Reference Hwang, Shen, Chu, Yui, Varela and Iizuka2015), described as Ca silico-phosphate or Si apatite in the literature (Kaneda et al., Reference Kaneda, Mikouchi, Saito, Sugiyama, Ohsumi, Mukai, Osaka, Miyata, Nakai, Kasama, Chikami and Miyamoto2001; Mittlefehldt et al., Reference Mittlefehldt, Killgore and Lee2002; Mikouchi et al., Reference Mikouchi, Kaneda, Miyamoto, Sugiyama and Ohsumi2001, Reference Mikouchi, Sugiyama, Kato, Yamaguchi, Koizumi and Kaneda2010, Reference Mikouchi, Sugiyama, Satake and Amelin2011; Kurat et al., Reference Kurat, Varela, Brandstätter, Weckwerth, Clayton, Weber, Schultz, Wäsch and Nazarov2004); new mineral matyhite (Ma) (Hwang et al., Reference Hwang, Shen, Chu, Yui, Varela and Iizuka2016b), described as Ca phosphate in the literature (Mittlefehldt et al., Reference Mittlefehldt, Killgore and Lee2002; Kurat et al., Reference Kurat, Varela, Brandstätter, Weckwerth, Clayton, Weber, Schultz, Wäsch and Nazarov2004); less commonly Na-bearing anorthite; and some unknown phases and voids (Fig. S1a,b). Besides the coarse Fa–Kir overgrowth over the forsterite core (Fig. S1c; hereinafter referred to as Fa–Kir overgrowth) commonly reported in the literature (e.g. Mittlefehldt et al., Reference Mittlefehldt, Killgore and Lee2002; Kurat et al., Reference Kurat, Varela, Brandstätter, Weckwerth, Clayton, Weber, Schultz, Wäsch and Nazarov2004), there is another type of much finer symplectitic Fa–Kir intergrowth surrounding the resorbed Fe sulfide in the proximity of hedenbergite (Fig. S1d; hereinafter referred to as Fa–Kir symplectite). The representative compositions of the major phases in the D'Orbigny angrite are listed in Table S1 (Supplementary material – see below), and detailed petrographic descriptions and debate on the possible non-igneous origin of D'Orbigny can be found in Mittlefehldt et al. (Reference Mittlefehldt, Killgore and Lee2002), Kurat et al. (Reference Kurat, Varela, Brandstätter, Weckwerth, Clayton, Weber, Schultz, Wäsch and Nazarov2004) and Varela et al. (Reference Varela, Kurat, Zinner, Hoppe, Ntaflos and Nazarov2005).
Occurrences of tsangpoite and matyhite
Optical observations
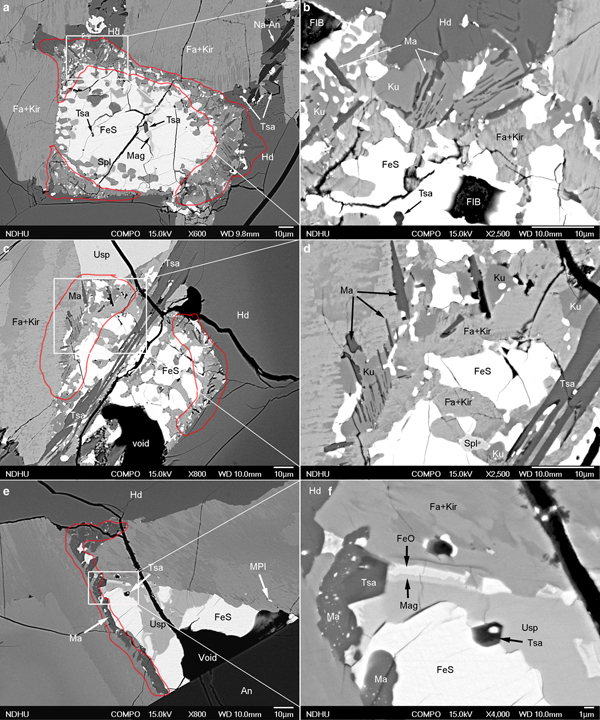
Tsangpoite and matyhite occur specifically in well-defined but separated domains associated with Fe sulfide in the proximity of the contact between Fa–Kir overgrowth/symplectite and hedenbergite (see Fig. S1). Because of the high abundance, larger crystal sizes, and oriented rod-like growth morphology, tsangpoite was readily discernible petrographically by the relief and colour of the phases. Optical microscopy observations showed that groups of subparallel tsangpoite crystals with greyish colour usually nucleated at Fe sulfide and grew tangentially to the wall of Fa–Kir overgrowths into the space currently filled by brownish hedenbergite (Fig. 1a,b). Further OM observations showed that tsangpoite crystals with hexagonal cross-sections on the petrographic thin section surface could usually be traced back to the resorbed Fe sulfide using the focus (Fig. S2). Such elongated tsangpoite crystals frequently contain a central tube filled with opaque minerals of FeS + Mag ± ferrite (bcc Fe) ± Fe–S–O phases (Fig. 1c), as confirmed by electron imaging and diffraction (not shown). By contrast, because of the lower abundance and smaller sizes, it was more difficult to recognise/image matyhite in OM analyses. Nevertheless, some observations did show that greyish matyhite plate-shaped crystals usually nucleated and grew from the Fa–Kir symplectitic margin next to Fe sulfide into hedenbergite (Fig. 1d).
Fig. 1. Group of subparallel tsangpoite crystals that nucleated at FeS and grew tangentially to the wall of the Fa–Kir overgrowth: (a) viewed transversely; and (b) viewed nearly end-on. (c) The constant presence of segmental FeS + magnetite + Fe along the central tube of tsangpoite; and (d) group of dendritical matyhite plates that grew from the Fa–Kir symplectite margin of FeS. Image details: transmitted light, parallel Nicols.
SEM-BSE observations
Observations from BSE analysis further showed that tsangpoite crystals either occur within Fe sulfide or Fa–Kir symplectite after Fe sulfide, or are currently embedded within the hedenbergite domain close to resorbed Fe sulfide. In the ‘intact’ Fe sulfide not in direct contact with hedenbergite, between either anorthite and the Fa–Kir overgrowth (Fig. 2a; Fa–Kir overgrowth not shown), or anorthite and ulvöspinel (Fig. 2b), or the impinged Fa–Kir overgrowths (Fig. 2c), tsangpoite crystals ~1–5 µm in diameter with hexagonal cross-section occur exclusively in the local partial-melting-like domain subjected to partial replacement of Fe sulfide by Mag + wüstite + Tsa ± celsian ± Al–Ti–Fe Spl (24–39 Usp, 24–31 Hc, 21–30 Mag; TEM-EDX; abbreviations according to Whitney and Evans, Reference Whitney and Evans2010) (Fig. 2a–c).
Fig. 2. Back-scatter electron micrographs showing the oxidation assemblage: FeS => Tsa + Mag + FeO ± celsian ± Al–Ti–Fe Spl within the ‘intact’ FeS at (a) contact of anorthite and Fa + Kir overgrowths (Fa + Kir overgrowths not shown), (b) contact of anorthite and ulvöspinel or (c) the impinged Fa–Kir overgrowths, and abundant tsangpoite crystals within (d,e) residue FeS, or (d–h) the Fa + Kir symplectite after FeS in the proximity of hedenbergite, as well as (g–i) the dissociation/decomposition of the Fa–Kir symplectite and the formation of well-defined tsangpoite domains in front of resorbed FeS. Note the partial melting-like Mag + FeO domain in (a–c), the bimodal size distribution of tsangpoite in (d–i), the tsangpoite + kuratite association in (g), as well as the dissociation-like feature of resorbed FeS in (h,i). Abbreviations according to Whitney and Evans (Reference Whitney and Evans2010); Tsa – tsangpoite.
In contrast to rare tsangpoite at the aforementioned domains, abundant tsangpoite crystals occur at the resorbed Fe sulfide domains currently in contact with hedenbergite (Fig. 2d–i). At such domains, tsangpoite crystals reside within either residue Fe sulfide (Fig. 2d,e) or the Fa–Kir symplectite domain after Fe sulfide (Fig. 2e–h). The Fe sulfide crystal and the associated Fa–Kir symplectite appear to be relatively unstable and hence gradually dissociated/decomposed, thereby eventually exposing many tsangpoite crystals in a space of up to ~100–200 µm right in front of the resorbed Fe sulfide (Fig. 2g–i). Through-focus OM observations showed that many such tsangpoite crystals could be traced back to the decomposed Fe sulfide (e.g. Fig. 1a,b; Fig. S2). Tsangpoite crystals thus formed have an interesting size distribution. The large crystals (~10–20 µm) at the outermost apron away from the Fe sulfide core typically have irregular cross-sections (e.g. Fig. 2f–i) indicating rather high growth rates under high driving force. By contrast, the tsangpoite crystals close to or still within Fe sulfide or Fa–Kir symplectite are generally much smaller in size (~1–5 µm) and have a characteristic ‘imperfect’ hexagonal form by the {10![]() $\bar 1 $0} facets along with [0001]-oriented longitudinal grooves with rugged interiors, probably resulting from the matrix and/or solute hindrance on fast crystal growth (Figs 2e–h, 3a,b). In fact, tsangpoite tends to group as subparallel crystals aligned along the common [0001] growth direction to show similar cross-sections in the petrographic thin section (Fig. 3b,c). Besides the aforementioned tsangpoite with the ‘normal’ hexagonal crystal form, skeletal tsangpoite crystals, probably driven by the very high crystal growth rates, are also not uncommon (Fig. 3d). As noted in OM observations, the tsangpoite crystals that nucleated and grew from Fe sulfide frequently possess a central tube filled with opaque minerals (e.g. Fig. 1c). Such minerals, i.e. FeS + Mag ± ferrite ± Fe–S–O phases were also observed readily in SEM-BSE and EDX analyses of two subparallel tsangpoite crystals nucleated in an Fe sulfide, and grew outwards into the gap between two Fa–Kir overgrowths (Fig. 3e,f).
$\bar 1 $0} facets along with [0001]-oriented longitudinal grooves with rugged interiors, probably resulting from the matrix and/or solute hindrance on fast crystal growth (Figs 2e–h, 3a,b). In fact, tsangpoite tends to group as subparallel crystals aligned along the common [0001] growth direction to show similar cross-sections in the petrographic thin section (Fig. 3b,c). Besides the aforementioned tsangpoite with the ‘normal’ hexagonal crystal form, skeletal tsangpoite crystals, probably driven by the very high crystal growth rates, are also not uncommon (Fig. 3d). As noted in OM observations, the tsangpoite crystals that nucleated and grew from Fe sulfide frequently possess a central tube filled with opaque minerals (e.g. Fig. 1c). Such minerals, i.e. FeS + Mag ± ferrite ± Fe–S–O phases were also observed readily in SEM-BSE and EDX analyses of two subparallel tsangpoite crystals nucleated in an Fe sulfide, and grew outwards into the gap between two Fa–Kir overgrowths (Fig. 3e,f).
Fig. 3. Back-scatter electron micrographs showing: (a,b) imperfect hexagonal cross-section with longitudinal groove of tsangpoite in Fa–Kir symplectite; (b,c) groups of subparallel tsangpoite crystals with similar cross-section morphologies exposed in hedenbergite; (d) a highly defective skeletal tsangpoite crystal viewed transversally; and (e,f) two subparallel oblique tsangpoite crystals originated from the same FeS crystal with central tube filled by FeS + Mag + Fe.
Unlike tsangpoite, matyhite was not found in the relatively ‘intact’ Fe sulfide at the sample domains away from hedenbergite (e.g. Fig. 2a–c). Instead, matyhite crystals frequently nucleated/grew within the Fa–Kir symplectite after Fe sulfide (Fig. 4a,b), yielding dendritic matyhite enclosed within Fa–Kir symplectite (Fig. 4b). Matyhite with high abundance of tiny Fe sulfide droplets and/or spherical voids also occurs frequently at the areas of contact between Fa–Kir symplectite and hedenbergite (Fig. 4c). Similarly to the case of tsangpoite, the gradual dissociation/decomposition of resorbed Fe sulfide and associated Fa–Kir symplectite also accounts for the exposure of some dendritic matyhite plates in the current hedenbergite phase, as illustrated nicely by the micrographs in Fig. 4d–f.
Fig. 4. Back-scatter electron micrographs showing the formation of matyhite plates (a,b) within Fa–Kir symplectite; (c) tangential to the contact between Fa–Kir symplectite and hedenbergite; and (d–f) the dissolution/decomposition of Fa–Kir symplectite and the consequential exposure of dendritical matyhite plates in hedenbergite. Note the frequent inclusion of abundant FeS droplets and/or spherical voids in matyhite (c–f).
Tsangpoite and matyhite were also commonly distributed at the core and rim, respectively, in the same resorbed Fe sulfide crystals (Fig. 5a,c,e). For such occurrences, matyhite formed within Fa–Kir symplectite domains in contact with either hedenbergite or a Fa–Kir overgrowth (Fig. 5a,c). Groups of subparallel matyhite plates were frequently embedded within kuratite (e.g. Fig. 5b,d), albeit there are no specific crystallographic relationships in between based on electron diffraction (not shown). Although relatively rare, matyhite partly surrounding tsangpoite was noted, as shown in Fig. 5e,f. According to electron diffraction (not shown), there are no definite crystallographic relationships between such a tsangpoite–matyhite pair. Whereas the dendritic plates currently embedded within hedenbergite could grow to the size of ~30 µm (width) × ~5 µm (thick) (e.g. Fig. 4f), the matyhite dendritic plates within the Fa–Kir symplectite or kuratite are rather small, i.e. ~10 µm (width) × ~0.2 µm (thick) (e.g. Fig. 5b,d).
Fig. 5. Back-scatter electron micrographs showing the concurrent presence of (a–d) tsangpoite at the core and matyhite (outlined in red) at the rim of resorbed Fe sulfide; and (e,f) tsangpoite at the core and matyhite (outlined in red) at the areas of contact with Fa–Kir overgrowth of a partially altered Fe sulfide. The skeletal morphology of tsangpoite crystal was noted in (c,d) and the occasional contact between tsangpoite and post-dated matyhite was noted in (f).
Crystallography
Tsangpoite
On the basis of least-squares refinement of 19 d-spacing measurements from electron diffraction patterns (Table 1), tsangpoite was determined conclusively to have a hexagonal unit cell with a = 9.489(4) Å, c = 6.991(6) Å, V = 545.1(6) Å3 and Z = 2 for 12 oxygen atoms per formula unit, despite the absence of single-crystal XRD data because of cryshttp://www.icdd.com/al size restriction.
Table 1. Observed and calculated d spacings (Å) for tsangpoite and related phases.
1Quenched from 1500°C; 2high-temperature camera powder measurements at ~1500°C; 3powder diffraction files from the International Centre for Diffraction Data (http://www.icdd.com/).
*Duplicated data, not used for lattice parameters refinement; **reflection not consistent with α-Ca2SiO4 unit cell.
The strongest reflections are marked in bold; w – weak; st – strong; vw – very weak; m – moderate.
The systematic absence of reflections 000l with l = 2n + 1, as confirmed by tilting experiments away from the exact zone axis orientation to nullify double diffraction effects, indicates a 63 screw axis along c (Fig. 6a,b). The ‘non-omission’ of alternating columns of diffraction spots of {hh ![]() $\bar {2h} $l} or {h
$\bar {2h} $l} or {h ![]() $\bar h $0l} with l = 2n + 1 in <11
$\bar h $0l} with l = 2n + 1 in <11![]() $\bar {\rm 2} $0> (Fig. 6a) and <1
$\bar {\rm 2} $0> (Fig. 6a) and <1![]() $\bar 1 $00> (Fig. 6b) zone axis patterns further excludes the possibility of the presence of c glide planes parallel to c. The intensity symmetry of the <0001> zone axis pattern, e.g. 2
$\bar 1 $00> (Fig. 6b) zone axis patterns further excludes the possibility of the presence of c glide planes parallel to c. The intensity symmetry of the <0001> zone axis pattern, e.g. 2![]() $\bar {\rm 3} $10 > 1
$\bar {\rm 3} $10 > 1![]() $\bar {\rm 3} $20 (arrowed in Fig. 6c), further indicates that there are no 2-fold axes normal to the c axis. These considerations leave only P63/m and P63 as possible space groups for tsangpoite. In this regard, it is noted that XRD data from a relatively large calcium silico-phosphate crystal (tsangpoite) from the NWA angrite 4590 indicated a hexagonal P63/m structure with a = 9.479 and c = 6.97 Å (Mikouchi et al., Reference Mikouchi, Sugiyama, Satake and Amelin2011), in close agreement with the electron diffraction results presented here. Note that due to space-group specification, the <hkil > family may show different intensity symmetry, cf. Fig. S3a,b for the case of <
$\bar {\rm 3} $20 (arrowed in Fig. 6c), further indicates that there are no 2-fold axes normal to the c axis. These considerations leave only P63/m and P63 as possible space groups for tsangpoite. In this regard, it is noted that XRD data from a relatively large calcium silico-phosphate crystal (tsangpoite) from the NWA angrite 4590 indicated a hexagonal P63/m structure with a = 9.479 and c = 6.97 Å (Mikouchi et al., Reference Mikouchi, Sugiyama, Satake and Amelin2011), in close agreement with the electron diffraction results presented here. Note that due to space-group specification, the <hkil > family may show different intensity symmetry, cf. Fig. S3a,b for the case of <![]() $\bar 5 $140> and Fig. S3g,h for the case of <
$\bar 5 $140> and Fig. S3g,h for the case of <![]() $\bar 5 $14
$\bar 5 $14![]() $\bar 9 $>. The ten relatively strong reflections in order of decreasing d spacing (in Å) are:
$\bar 9 $>. The ten relatively strong reflections in order of decreasing d spacing (in Å) are: ![]() $\bar 1 \,\bar 1 $21 (3.94), 0002 (3.50), 2
$\bar 1 \,\bar 1 $21 (3.94), 0002 (3.50), 2![]() $\bar {\rm 3} 10\, (3.10),\, 2\bar {\rm 3} 11 \,\,{\rm and} \,\, 1\bar {\rm 3} 21\, (2.83), 1\bar {\rm 2} 1\bar {\rm 2}\, (2.82),\, 3\bar {\rm 3} 00 \,(2.74), 20\bar {\rm 2} \bar {\rm 2}$ (2.66),
$\bar {\rm 3} 10\, (3.10),\, 2\bar {\rm 3} 11 \,\,{\rm and} \,\, 1\bar {\rm 3} 21\, (2.83), 1\bar {\rm 2} 1\bar {\rm 2}\, (2.82),\, 3\bar {\rm 3} 00 \,(2.74), 20\bar {\rm 2} \bar {\rm 2}$ (2.66), ![]() $\bar 14$
$\bar 14$![]() $\bar {\rm 3} $0 (2.28),
$\bar {\rm 3} $0 (2.28), ![]() $\bar {\rm 2}\, \bar {\rm 2} $42 (1.97) and
$\bar {\rm 2}\, \bar {\rm 2} $42 (1.97) and ![]() $\bar 4 $402 (1.77) (Fig. S3). The d spacings, symmetry, lattice parameters, and unit-cell correspondence of tsangpoite, hydroxylapatite, as well as the high-temperature hexagonal solid-solution phase and the low-temperature intermediate phases in the system Ca2SiO4–Ca3(PO4)2 are compiled in Tables 1 and 2 for comparison (see Discussion).
$\bar 4 $402 (1.77) (Fig. S3). The d spacings, symmetry, lattice parameters, and unit-cell correspondence of tsangpoite, hydroxylapatite, as well as the high-temperature hexagonal solid-solution phase and the low-temperature intermediate phases in the system Ca2SiO4–Ca3(PO4)2 are compiled in Tables 1 and 2 for comparison (see Discussion).
Table 2. Lattice parameters of tsangpoite, α-Ca2SiO4, silico-phosphate and hydroxylapatite.
[1] Mikouchi et al. (Reference Mikouchi, Sugiyama, Satake and Amelin2011); [2] Yamaguchi et al. (Reference Yamaguchi, Ono, Kawamura and Soda1963), 1500°C; [3] Mumme et al. (Reference Mumme, Cranswick and Chakoumakos1996); [4] Fukuda et al. (Reference Fukuda, Maki, Ito and Miyake1997), impurity stabilised α-Ca2SiO4, quenched from 1500°C; [5] Widmer et al. (Reference Widmer, Gfeller and Armbruster2015), quenched from 1300°C, with diffuse flamite-like reflections; [6] Bredig (Reference Bredig1942; Reference Bredig1943); [7] Nurse et al. (Reference Nurse, Welch and Gutt1959), refined from 5 d spacings at ~1500°C; [8] Fukuda et al. (Reference Fukuda, Maki, Toyoda and Ito1993), impurity stabilised α-Ca2SiO4, quenched from 1400°C; [9] Bredig (Reference Bredig1943), impurity stabilised α-Ca2SiO4; [10] Saalfed and Klaska (Reference Saalfeld and Klaska1981); [11] Gfeller et al. (Reference Gfeller, Widmer, Krüger, Galuskin and Armbruster2015); [12] Widmer et al. (Reference Widmer, Gfeller and Armbruster2015); [13] Galuskin et al. (Reference Galuskin, Galuskina, Gfeller, Krüger, Kusz, Vapnik, Dulski and Dzierżanowski2016); and [14] Marchat et al. (Reference Marchat, Zymelka, Coelho, Gremillard, Joly-Pottuz, Babonneau, Esnouf, Chevalier and Bernache-Assollant2013).
Fig. 6. The representative electron-diffraction patterns of (a–c) tsangpoite, and (d–f) matyhite in the principal zone axes.
Matyhite
Based on the least-squares refinement of 20 d-spacing measurements from electron diffraction patterns (Table 3), matyhite was determined conclusively to have a trigonal unit cell with a = 10.456(7) Å, c = 37.408(34) Å, V = 3541.6(4.8) Å3 and Z = 6 for 28 oxygen atoms per formula unit. The set of d spacings and the refined unit-cell parameters of matyhite in the present study are comparable to those of other merrillite-group minerals, whitlockite and synthetic β-TCP (Tables 3 and 4). The systematic absence of reflections 000l with l ≠ 6n (Fig. 6d), absence of reflections with –h + k+l ≠ 3n (Fig. 6d), as well as the absence of reflections hhl with l ≠ 3n (Fig. 6e) indicate that matyhite has R3c space group, which is the common space group for whitlockite/merrillite-group minerals. The nine relatively strong reflections in order of decreasing d spacing (in Å) are: 0![]() $\bar 1 $14 (6.52), 2
$\bar 1 $14 (6.52), 2![]() $\bar 1 \bar 1 $0 (5.24), 0
$\bar 1 \bar 1 $0 (5.24), 0![]() ${\bar 1} 1 .10 (3.46), 3{\bar 1} \bar {\rm 2} $4 (3.21), 30
${\bar 1} 1 .10 (3.46), 3{\bar 1} \bar {\rm 2} $4 (3.21), 30![]() $\bar {\rm 3} $0 (3.02), 0
$\bar {\rm 3} $0 (3.02), 0![]() $\bar {\rm 2} $2.
$\bar {\rm 2} $2.![]() $\bar {{\rm 10}} $ (2.88),
$\bar {{\rm 10}} $ (2.88), ![]() $\bar {\rm 3} $128 (2.75), 4
$\bar {\rm 3} $128 (2.75), 4![]() $\bar {\rm 2} \bar {\rm 2} $0 (2.62) and 3
$\bar {\rm 2} \bar {\rm 2} $0 (2.62) and 3![]() $\bar 1 \bar {\rm 2} $.10 (2.53) (Fig. S4).
$\bar 1 \bar {\rm 2} $.10 (2.53) (Fig. S4).
Table 3. Observed and calculated d spacings (Å) for matyhite* and related phases.
*The strongest reflections are marked in bold.
[1] Suizho meteorite (Xie et al., Reference Xie, Yang, Gu and Downs2015), American Mineralogist Crystal Structure Database (amcsd)-0020386; [2] Angra dos Reis (Dowty, Reference Dowty1977), amcsd-0018313; [3] Shergotty meteorite (Britvin et al., Reference Britvin, Krivovichev and Armbruster2016). [4] Lunar rock, Fra Mauro Formation (Hughes et al., Reference Hughes, Jolliff and Gunter2006), amcsd-0004238; [5] Palermo Quarry (Gopal and Calvo, Reference Gopal and Calvo1972), amcsd-0000445; and [6] Dickens et al. (Reference Dickens, Schroeder and Brown1974), JCPDS file 70-2065 (powder diffraction files from the International Centre for Diffraction Data, http://www.icdd.com/).
Table 4. Lattice parameters of merrillite-group minerals, whitlockite, and synthetic β-tricalcium phosphate.
[1] Suizho meteorite (Xie et al., Reference Xie, Yang, Gu and Downs2015); [2] Angra dos Reis (Dowty, Reference Dowty1977); [3] Shergotty meteorite (Britvin et al., Reference Britvin, Krivovichev and Armbruster2016); [4] Lunar rock, Fra Mauro Formation (Hughes et al., Reference Hughes, Jolliff and Gunter2006); [5] Palermo Quarry (Gopal and Calvo, Reference Gopal and Calvo1972); [6] Dickens et al. (Reference Dickens, Schroeder and Brown1974).
TEM imaging
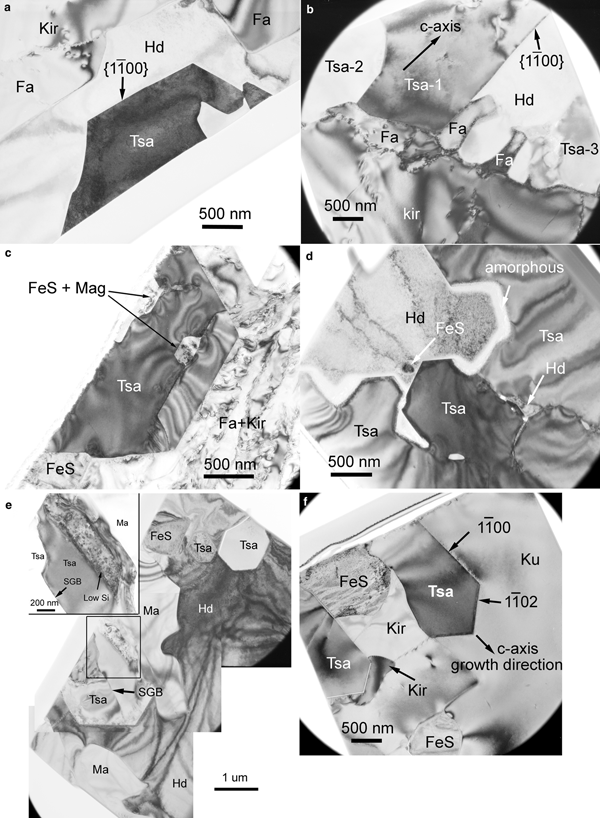
Transmission electron microscopy bright field (TEM BF) images of tsangpoite within hedenbergite or Fa–Kir symplectite compiled in Fig. 7 show the characteristic imperfect [0001] cross-sections of tsangpoite (Fig. 7a), the frequent association of tsangpoite and fayalite (Fig. 7a,b), Fe sulfide + Mag at the central tube of tsangpoite (Fig. 7c), as well as the interesting, grain-coalescence-like zigzag outline of the interior of an open, [0001]-oriented groove of tsangpoite similar to that shown in Fig. 3a,b (Fig. 7d). Several occasional crystallographic orientation relationships were found between tsangpoite and the olivine matrix, e.g. <0001>Tsa ~//~ <100>Ol and {1![]() $\bar 1 $00}Tsa ~//~ {011}Ol for the pair in Fig. 7a (tsangpoite appears to be detached from the olivine–hedenbergite boundary, leaving a negative crystal shape of olivine), or <11
$\bar 1 $00}Tsa ~//~ {011}Ol for the pair in Fig. 7a (tsangpoite appears to be detached from the olivine–hedenbergite boundary, leaving a negative crystal shape of olivine), or <11![]() $\bar {\rm 2} $6>Tsa ~//~ <210>Ol and {1
$\bar {\rm 2} $6>Tsa ~//~ <210>Ol and {1![]() $\bar 1 $00}Tsa ~//~ {001}Ol for the pair in Fig. 7c, or <11
$\bar 1 $00}Tsa ~//~ {001}Ol for the pair in Fig. 7c, or <11![]() $\bar {\rm 2} $0>Tsa ~//~ <014>Ol and {1
$\bar {\rm 2} $0>Tsa ~//~ <014>Ol and {1![]() $\bar 1 $00}Tsa ~//~ {34
$\bar 1 $00}Tsa ~//~ {34![]() $\bar 1 $}Ol (not shown), or <11
$\bar 1 $}Ol (not shown), or <11![]() $\bar {\rm 2} $3>Tsa ~//~ <110>Ol and {1
$\bar {\rm 2} $3>Tsa ~//~ <110>Ol and {1![]() $\bar 1 $00}Tsa ~//~ {001}Ol (not shown), or <11
$\bar 1 $00}Tsa ~//~ {001}Ol (not shown), or <11![]() $\bar {\rm 2} $3>Tsa ~//~ <112>Ol and {1
$\bar {\rm 2} $3>Tsa ~//~ <112>Ol and {1![]() $\bar 1 $00}Tsa ~//~ {1
$\bar 1 $00}Tsa ~//~ {1![]() $\bar 1 $0}Ol (not shown).
$\bar 1 $0}Ol (not shown).
Fig. 7. TEM micrographs of tsangpoite showing (a) imperfect hexagonal [0001] cross-sections, (a,b) the constant presence of fayalite at the contact between tsangpoite and Fa + Kir overgrowth, (c) the presence of magnetite and FeS at the central tube, (d) the coalescence-like microstructure within the open [0001]-oriented groove filled by FeS + hedenbergite, as well as the association of (e) tsangpoite + matyhite (TEM section from Fig. 5f) and (f) tsangpoite + kuratite (TEM section from Fig. 2g). The two branches of tsangpoite grown around FeS in (f) are facetted by {1![]() $\bar 1 $00} and {1
$\bar 1 $00} and {1![]() $\bar 1 $02} planes. The Si-depleted thin boundary layer between tsangpoite and matyhite is shown in insert of (e).
$\bar 1 $02} planes. The Si-depleted thin boundary layer between tsangpoite and matyhite is shown in insert of (e).
Although quite rare, as noted from SEM analyses, a tsangpoite + matyhite association was observed occasionally (see Fig. 5f). TEM BF images show the phase distribution/arrangement of tsangpoite with hexagonal cross-section, matyhite with tabular cross-section and Fe sulfide, as well as the surrounding hedenbergite matrix in one TEM thin section prepared selectively from the tsangpoite + matyhite association in Fig. 5f (Fig. 7e). Electron diffraction showed that the three tsangpoite crystals in this thin section are nearly parallel to each other along their c axis (<3–5° off), consistent with the SEM observations. A subgrain boundary is not common, but was found occasionally within one tsangpoite crystal (Fig. 7e). Despite the fact that tsangpoite appears to be oriented within the hedenbergite matrix, as also observed in SEM micrographs, there are no specific crystallographic orientation relationships between them, according to an electron diffraction study (not shown). At the contact between tsangpoite and matyhite, a thin layer depleted in Si (compared to tsangpoite) was noted by TEM-EDX analysis (insert in Fig. 7e). Figure 7f is a TEM BF image of another focused ion beam thin section prepared selectively from the tsangpoite–kuratite association in Fig. 2g showing the association of tsangpoite + kuratite in Fa–Kir symplectite (see Fig. 2g). Note that the tiny tsangpoite crystal branching into two halves and facetted by {1![]() $\bar 1 $00} and {1
$\bar 1 $00} and {1![]() $\bar 1 $02} planes probably nucleated at the central Fe sulfide and grew along the crystallographic c axis. There is no definite crystallographic orientation relationship between tsangpoite and kuratite, according to electron diffraction.
$\bar 1 $02} planes probably nucleated at the central Fe sulfide and grew along the crystallographic c axis. There is no definite crystallographic orientation relationship between tsangpoite and kuratite, according to electron diffraction.
The results from TEM BF imaging and electron diffraction showed that the matyhite plates within a dendritic bundle in hedenbergite usually have approximately similar crystallographic orientation and habit in space (up to several degrees off). The majority of thick matyhite plates within the hedenbergite analysed are ~(0001) plates with segments of flat (0001) facets (Fig. 8a), although minor ~{11![]() $\bar 2 $0} plates were also observed (Fig. 8b). The presence of abundant voids of various morphologies in some matyhite plates within hedenbergite can be seen clearly in Fig. 8b,d (arrowed). As for the thin matyhite plates within kuratite, TEM imaging showed that a dendritic bundle of thin matyhite plates could be embedded by multiple kuratite crystals (Fig. 8c) or by a single kuratite crystal (Fig. 8d). The matyhite plates could be flat (e.g. Fig. 8c) or wavy (e.g. Fig. 8d), and the mis-orientations in-between (up to ~30°) are generally larger than that in hedenbergite. Minor hedenbergite in good crystallographic orientation relationships to kuratite, as well as Fe sulfide in random orientations, were also frequently noted in such domains (e.g. Fig. 8c). The matyhite crystals in kuratite could be either ~{11
$\bar 2 $0} plates were also observed (Fig. 8b). The presence of abundant voids of various morphologies in some matyhite plates within hedenbergite can be seen clearly in Fig. 8b,d (arrowed). As for the thin matyhite plates within kuratite, TEM imaging showed that a dendritic bundle of thin matyhite plates could be embedded by multiple kuratite crystals (Fig. 8c) or by a single kuratite crystal (Fig. 8d). The matyhite plates could be flat (e.g. Fig. 8c) or wavy (e.g. Fig. 8d), and the mis-orientations in-between (up to ~30°) are generally larger than that in hedenbergite. Minor hedenbergite in good crystallographic orientation relationships to kuratite, as well as Fe sulfide in random orientations, were also frequently noted in such domains (e.g. Fig. 8c). The matyhite crystals in kuratite could be either ~{11![]() $\bar 2 $6} plates (Fig. 8c,d), or ~{10
$\bar 2 $6} plates (Fig. 8c,d), or ~{10![]() $\bar 1 $4} plates (Fig. 8e,f). The bundle of thin ~{10
$\bar 1 $4} plates (Fig. 8e,f). The bundle of thin ~{10![]() $\bar 1 $4} plates within fine Fa–Kir symplectite also contain many sub-grain boundaries (Fig. 8f), in addition to the irregular voids (Fig. 8f).
$\bar 1 $4} plates within fine Fa–Kir symplectite also contain many sub-grain boundaries (Fig. 8f), in addition to the irregular voids (Fig. 8f).
Fig. 8. TEM micrographs of matyhite dendritic plates showing: (a) the most commonly observed thick {0001} plates within hedenbergite; (b) the less commonly observed thick {11![]() $\bar {\rm 2} $0} plates with abundant voids of various shapes within hedenbergite; (c) a bundle of thin ~{11
$\bar {\rm 2} $0} plates with abundant voids of various shapes within hedenbergite; (c) a bundle of thin ~{11![]() $\bar {\rm 2} $6} plates with flat surfaces and associated hedenbergite + FeS embedded by multiple kuratite crystals; (d) a bundle of thin ~{11
$\bar {\rm 2} $6} plates with flat surfaces and associated hedenbergite + FeS embedded by multiple kuratite crystals; (d) a bundle of thin ~{11![]() $\bar {\rm 2} $6} plates with very rugged surfaces and surface voids within a single kuratite crystal; (e) a thin ~{10
$\bar {\rm 2} $6} plates with very rugged surfaces and surface voids within a single kuratite crystal; (e) a thin ~{10![]() $\bar 1 $4} plate of matyhite with subgrain structure enclosed by kuratite and attached to olivine at the edge; and (f) a bundle of thin ~{10
$\bar 1 $4} plate of matyhite with subgrain structure enclosed by kuratite and attached to olivine at the edge; and (f) a bundle of thin ~{10![]() $\bar 1 $4} plates with subgrain structure and voids within Fa + Kir symplectite. Some voids in (b,d,f) are indicated by arrows.
$\bar 1 $4} plates with subgrain structure and voids within Fa + Kir symplectite. Some voids in (b,d,f) are indicated by arrows.
Raman spectroscopy
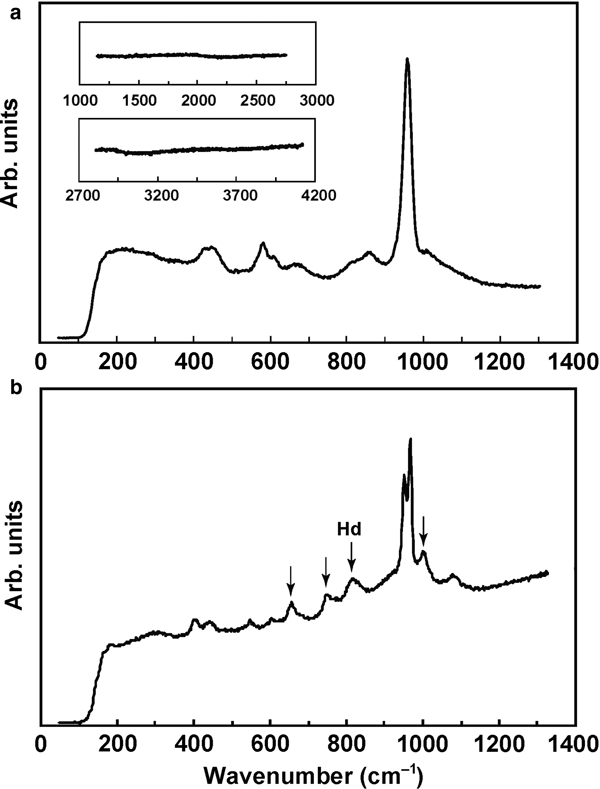
The Raman spectrum of tsangpoite shows six bands: 437–448 and 460–465 (ν2 PΟ4), 591–601 (ν4 PΟ4), 867–871 (Si–O stretching), 959–961 (ν1 PΟ4, strongest) and ~1019 (ν3 PΟ4), as well as a weak and broad band at ~630–700 cm–1 (Fig. 9a; see also Mikouchi et al., Reference Mikouchi, Sugiyama, Kato, Yamaguchi, Koizumi and Kaneda2010). This spectrum basically resembles that of nagelschmidtite (402–439, 587, 642–647, 857, 961–963, 1058 and 1084 cm–1; Lugo et al., Reference Lugo, Mazón, Baudin and de Aza2015; Rabadan-Ros et al., Reference Rabadan-Ros, Velásquez, Meseguer-Olmo and de Aza2016), silicocarnotite (418, 593, ~634 or ~700 (in 2 analyses), 854, 959–963 and 1000–1150 cm–1; Serena et al., Reference Serena, Sainz and Caballero2014; Reference Serena, Caballero, de Aza and Sainz2015; 397, 474, 557, 584, ~626–734 with a series of vibrations, 850, 957 and ~1000–1100 cm–1; Galuskin et al., Reference Galuskin, Galuskina, Gfeller, Krüger, Kusz, Vapnik, Dulski and Dzierżanowski2016), and probably Si-substituted hydroxylapatite with the ~630–700 cm–1 band missing (~450, ~600, 847, 954–972 and ~1050 cm–1; Gomes et al., Reference Gomes, Nedelec, Jallot and Sheptyakov2011). The low intensity band at ~630–700 cm–1 of tsangpoite probably has a similar cause as the un-assigned ~645 cm–1 band of nagelschmidtite in Lugo et al. (Reference Lugo, Mazón, Baudin and de Aza2015) and Rabadan-Ros et al. (Reference Rabadan-Ros, Velásquez, Meseguer-Olmo and de Aza2016), the ~700 cm–1 band of silicocarnotite in Serena et al. (Reference Serena, Sainz and Caballero2014) considered to be related to Si–O–Si bending modes (e.g. Ibáñez et al., Reference Ibáñez, Artús, Cuscó, López, Menéndez and Andrade2007), or the ~626–734 cm–1 band of silicocarnotite which has a series of vibrations with the ‘strongest’ at 640 cm–1 attributed to ν4 PO4 by Galuskin et al. (Reference Galuskin, Galuskina, Gfeller, Krüger, Kusz, Vapnik, Dulski and Dzierżanowski2016). The lack of a Raman peak at ~2000, ~3000 or ~3500 cm–1, characteristic of OH vibrations, indicates that tsangpoite is OH free. Investigations using micro Fourier-transform infrared of a relatively large calcium silico-phosphate crystal (tsangpoite) from the NWA angrite 4590 also showed no OH content (Mikouchi et al., Reference Mikouchi, Sugiyama, Satake and Amelin2011). The broader and weaker SiO4 vibrations at ~850 cm–1, and probably also 630–700 cm–1, of tsangpoite compared to that of nagelschmidtite and silicocarnotite could be due to structural vacancies and aliovalent cations accompanying the substitution of Si for P for volume and charge compensation to shift vibration frequencies.
Fig. 9. Raman spectra of (a) tsangpoite and (b) matyhite with an additional peak due to the hedenbergite (Hd) matrix.
The Raman spectrum of matyhite shows several scattering bands near 414 (ν2 PΟ4), 458 (ν2 PΟ4), 573 (ν4 PΟ4), 629 (ν4 PΟ4), 953 (ν1 PΟ4), 959 (ν1 PΟ4) and 1097 (ν3 PΟ4) cm–1 (Fig. 9b), resembling that of synthetic merrillite or β-tricalcium phosphate (de Aza et al., Reference de Aza, Santos, Pazo, de Aza, Cuscó and Artús1997, Mikouchi et al., Reference Mikouchi, Kaneda, Miyamoto, Sugiyama and Ohsumi2001; Jolliff et al., Reference Jolliff, Hughes, Freeman and Zeigler2006; Jillavenkatesa and Condrate Sr., Reference Jillavenkatesa and Condrate1998). The lack of a Raman peak at ~3500 cm–1 (not shown), characteristic of OH vibration, demonstrates that matyhite is OH free. The 662, 772, 840 and 1008 cm–1 bands (arrowed) in Fig. 9b are due to the hedenbergite matrix.
Composition
Tsangpoite
Eight analyses from four tsangpoite crystals and their averages are given in Table 5. Having similar reflection data to the α-Ca2SiO4-derived structure or apatite structure (see Discussion), tsangpoite lattice sites can be assigned by the formula M 10T 6O24 of silico-phosphate in the binary Ca2SiO4–Ca3(PO4)2 system, or the formula M 10T 6O24X 2 of the apatite structure where the tetrahedral T-site atoms are P + Si + S = 6(0.05) and other cations are at octahedral M sites. The empirical formula based on 6 P + Si + S and 24 O + F + Cl or 24 O, with rounding errors, is then (Ca8.07□0.84Fe3+0.75Ti0.20Al0.06REE 0.02Sr0.02Y0.01Cr0.01Ni0.01Zn0.01)Σ10.0[(P3.99Si1.97S0.06)Σ6.02(O23.72F0.23Cl0.04)Σ23.99] (Table 6), or (Ca8.16Fe3+0.76□0.74Ti0.20Al0.06REE 0.02Sr0.02Y0.01Cr0.01Ni0.01Zn0.01)Σ10.0[(P4.04Si1.99S0.06)Σ6.09O24](□1.73F0.23Cl0.04) (not shown in Table 6) where REE = rare-earth elements. The absence of H2O and CO2 was confirmed by Raman spectroscopy. The simplified formula is Ca5(PO4)2(SiO4), the same as the terrestrial orthorhombic mineral silicocarnotite (Galuskin et al., Reference Galuskin, Galuskina, Gfeller, Krüger, Kusz, Vapnik, Dulski and Dzierżanowski2016).
Table 5. Representative compositions (wt.%) of tsangpoite from the D'Orbigny angrite
‘–’ = not detected
Electron microprobe analytical data for the unknown mineral referred to as silico-phosphate or calcium silico-phosphate in various volcanic and plutonic angrites have been reported by Kaneda et al. (Reference Kaneda, Mikouchi, Saito, Sugiyama, Ohsumi, Mukai, Osaka, Miyata, Nakai, Kasama, Chikami and Miyamoto2001), Mittlefehldt et al. (Reference Mittlefehldt, Killgore and Lee2002) and Kurat et al. (Reference Kurat, Varela, Brandstätter, Weckwerth, Clayton, Weber, Schultz, Wäsch and Nazarov2004) for D'Orbigny, Jambon et al. (Reference Jambon, Boudouma, Fonteilles, Le Guillou, Badia and Barrat2008) for NWA 1670, Mikouchi et al. (Reference Mikouchi, Sugiyama, Satake and Amelin2011) for NWA 4590, and Warren and Davis (Reference Warren and Davis1995) and Mikouchi et al. (Reference Mikouchi, Sugiyama, Kato, Yamaguchi, Koizumi and Kaneda2010) for Asuka 881371 (Table 6). Except for the lower formula weights probably due to the omission of trace heavy elements (mainly Sr and REE) in analyses, the calculated empirical formula for other measurements from D'Orbigny (Table 6; Mittlefehldt et al., Reference Mittlefehldt, Killgore and Lee2002; Kurat et al., Reference Kurat, Varela, Brandstätter, Weckwerth, Clayton, Weber, Schultz, Wäsch and Nazarov2004; Kaneda et al., Reference Kaneda, Mikouchi, Saito, Sugiyama, Ohsumi, Mukai, Osaka, Miyata, Nakai, Kasama, Chikami and Miyamoto2001) are quite similar to that for tsangpoite in the present study. The 2nd datum of Mittlefehldt et al. (Reference Mittlefehldt, Killgore and Lee2002) with Si/P = 1 and the 4th datum with a much larger formula weight (985.6 g/mole) in Table 6 are probably due to different phase(s) or analytical artifacts. As for Ca silico-phosphates from other angrites (Table 6; Asuka 881371, Warren and Davis, Reference Warren and Davis1995, Mikouchi et al., Reference Mikouchi, Sugiyama, Kato, Yamaguchi, Koizumi and Kaneda2010; NWA 1670, Jambon et al., Reference Jambon, Boudouma, Fonteilles, Le Guillou, Badia and Barrat2008; NWA 4590, Mikouchi et al., Reference Mikouchi, Sugiyama, Satake and Amelin2011), except for the one measurement in NWA 1670 with lower Si/P = 0.23 and Fe3+/Fe = 60% (Jambon et al., Reference Jambon, Boudouma, Fonteilles, Le Guillou, Badia and Barrat2008), the calculated formula are also closely similar to that of tsangpoite. It is clear that this unknown Ca silico-phosphate mineral commonly present in various angrites, in all probability, is tsangpoite with variable Si/P ratios and with Fe mainly in 3+ charge as was also confirmed by micro-XANES measurements on tsangpoite from NWA 4590 (Mikouchi et al., Reference Mikouchi, Sugiyama, Satake and Amelin2011). Excluding the 2nd and 4th data of Mittlefehldt et al. (Reference Mittlefehldt, Killgore and Lee2002) in Table 6, an ~4–10% cation vacancy at the M site is noted for tsangpoite in various angrites. The unexpected presence of such vacancies, along with the variable Si/P ratio (0.23–0.67), explains the long existing uncertainty regarding the stoichiometry of the commonly present minor calcium silico-phosphate phase(s) in angrites; its composition was considered to be similar but not identical to nagelschmidtite Ca7(SiO4)2(PO4)2 or silicocarnotite Ca5(PO4)2(SiO4) (e.g. Mikouchi et al., Reference Mikouchi, Sugiyama, Kato, Yamaguchi, Koizumi and Kaneda2010) (see Discussion).
Table 6. Comparison of compositions (wt.%) of tsangpoite from this work and the literature in various angrites.
[1] Mittlefehldt et al. (Reference Mittlefehldt, Killgore and Lee2002), (Ca8.44Fe+30.68Ti0.17Al0.05Mn0.01□0.65)Σ10.0[(P4.07Si1.97)Σ6.04O24]; [2] Kurat et al. (Reference Kurat, Varela, Brandstätter, Weckwerth, Clayton, Weber, Schultz, Wäsch and Nazarov2004), (Ca8.20Fe+30.66Ti0.21Al0.04Mn0.01Mg0.01□0.87)Σ10.0[(P4.11Si2.02)Σ6.13O24]; [3] Kaneda et al. (Reference Kaneda, Mikouchi, Saito, Sugiyama, Ohsumi, Mukai, Osaka, Miyata, Nakai, Kasama, Chikami and Miyamoto2001), (Ca8.32Fe+30.65Ti0.20Al0.04Mn0.01□0.78)Σ10.0[(P4.13Si1.96)Σ6.09O24]; [4] Jambon et al. (Reference Jambon, Boudouma, Fonteilles, Le Guillou, Badia and Barrat2008); [5] Warren and Davis (Reference Warren and Davis1995), (Ca8.01Fe+30.70Ti0.21Al0.06Mn0.01Ni0.01□1.0)Σ10.0[(P4.06Si2.13)Σ6.19O24]; [6] Mikouchi et al. (Reference Mikouchi, Sugiyama, Satake and Amelin2011), (Ca8.78Fe+30.34Ti0.08Al0.08Mn0.01□0.71)Σ10.0[(P4.64Si1.41)Σ6.05O24]; [7] Mikouchi et al. (Reference Mikouchi, Sugiyama, Kato, Yamaguchi, Koizumi and Kaneda2010), (Ca8.49Fe+30.64Ti0.18Al0.04Mn0.01□0.68)Σ10.0[(P3.99Si1.88S0.05)Σ5.92(O23.46F0.51Cl0.08)Σ24.05]; [8] this work, see Table 5.
‘–’ = not detected
Matyhite
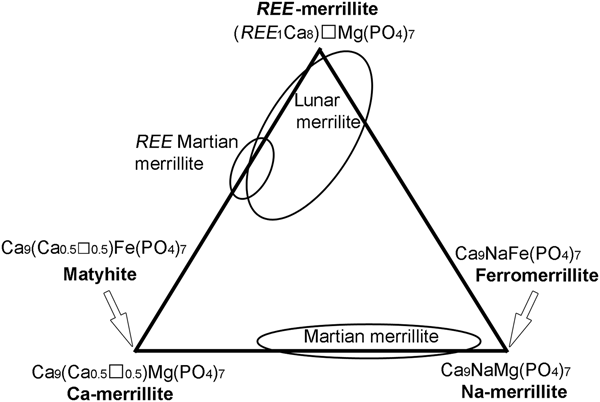
Due to the tiny sizes of matyhite plates, i.e. barely > 5 µm thick, it was difficult, if not impossible, to avoid interference from the matrix phase during EPMA. As the predominant hedenbergite matrix contains ~7.7 Si atoms and ~4.7 Ca atoms per 28 oxygens, it was noticed quickly that matyhite chemical data influenced by hedenbergite would readily show the sum of P + Si > 7 atoms per 28 oxygen atoms, and have the concurrent decrease in Ca. Based on these considerations, 18 analyses of matyhite with the sum of P + Si = 6.97–7.0 atoms per 28 oxygen atoms were considered acceptable and are compiled in Table 7. The lattice sites can then be assigned based on the merrillite formula (Ca9–x–z[Y,REE]x+z)Σ9Na1–x(Mg,Fe,Mn)1(P7–zSiz)Σ7O28, as outlined in Shearer et al. (Reference Shearer, Burger, Papike, McCubbin and Bell2015) where T-site cations are P + Si = 7(0.05) for the acceptable analyses and the octahedral M-site cations in the decreasing order of Al→Ti→Fe3+→Mg→Zn→Fe2+→Mn are a total of 1. The remaining cations are assigned to the 8-fold coordinated Ca site in the order Fe2+→Mn→Sr→REE, Y→Ca. This site is filled with 9 cations, and the excess Ca is then assigned to the Na site, along with Na and K. The empirical formula (atoms per formula unit) of the average analytical results, based on 28 oxygen atoms and 7 P + Si, is (Ca8.91Sr0.05REE 0.04)Σ9.0(□0.52Ca0.42Na0.05K0.01)Σ1.0(Fe2+0.68Fe3+0.26Al0.02Ti0.01Mn0.01Zn0.01)Σ0.99(P6.75Si0.26)Σ7.01O28.02 (with traces of Mg, Ni, Cl and S) (Table 8). The matyhite with the simplified formula Ca9(Ca0.5ο0.5)Fe(PO4)7 having the Na site (6-fold coordinated irregular octahedron) half empty and half occupied by Ca and the Mg site (6-fold coordinated regular octahedron) fully occupied by Fe, is in fact the Fe-analogue of ‘Ca-merrillite’ Ca9(Ca0.5□0.5)Mg(PO4)7 found in the Angra Dos Reis meteorite (Dowty, Reference Dowty1977) and was considered a new species in the merrillite group following the Criteria for New Mineral Species as detailed in Nickel and Grice (Reference Nickel and Grice1998). The calculated empirical formula of two Fe-rich merrillites from Martian meteorites (Shearer et al., Reference Shearer, Burger, Papike, McCubbin and Bell2015), and two occasional merrillite minerals from plutonic angrites (Prinz et al., Reference Prinz, Keil, Hlava, Berkley, Gomes and Curvello1977; McKay et al., Reference McKay, Lindstrom, Yang and Wagstaff1988) are included in Table 8 for comparison.
Table 7. Representative compositions (wt.%) of matyhite from the D'Orbigny angrite.
‘–’ = not detected
Table 8. Comparison of compositions (wt.%) of matyhite in various angrites from this work and the literature.
[1] Martian meteorites (Shearer et al., Reference Shearer, Burger, Papike, McCubbin and Bell2015), Ca9(□0.49Ca0.26Na0.25)Σ1.0(Fe2+0.91Mn0.04Al0.01Mg0.25)Σ1.21(P6.94Si0.03)Σ6.97O28 or Ca9(□0.69 Na0.17Ca0.14)Σ1.0(Fe2+0.57Fe3+0.38Mg0.10Mn0.04Ti0.01)Σ1.09(P6.98Si0.02)Σ7.0O28; [2] plutonic angrite, including 0.01% Y2O3 and 0.25% Ce2O3 (Prinz et al., Reference Prinz, Keil, Hlava, Berkley, Gomes and Curvello1977), Ca9(Ca0.52Na0.24□0.24)Σ1.0(Mg0.76Fe2+0.19)Σ0.95(P6.87Si0.12)Σ6.99O28; [3] plutonic angrite (Mckay et al., Reference McKay, Lindstrom, Yang and Wagstaff1988), Ca9(Ca0.77□0.23)Σ1.0(Mg0.72Fe2+0.24Al0.01Mn0.01)Σ0.98(P6.80Si0.13)Σ6.92O28; [4] this work, see Table 7.
‘–’ = not detected
Discussion
Relationship with other species
Tsangpoite
The comparable sets of unit-cell parameters, d spacings, strong reflections of tsangpoite, hydroxylapatite, and the high-temperature silico-phosphates in the binary Ca2SiO4–Ca3(PO4)2 system imply that tsangpoite could be related structurally to either apatite or the high-temperature hexagonal silico-phosphates analogous to the end-member α-Ca2SiO4 (Tables 1 and 2). In synthetic experiments of silicate-substituted hydroxylapatite at ~1000–1200°C; i.e. Ca10(PO4)6–y(SiO4)y(OH)2–y, the maximum limit of Si substitution into the hexagonal apatite lattice was found to be y = 1–1.2 (Gomes et al., Reference Gomes, Nedelec, Jallot and Sheptyakov2011; Marchat et al., Reference Marchat, Zymelka, Coelho, Gremillard, Joly-Pottuz, Babonneau, Esnouf, Chevalier and Bernache-Assollant2013). To maintain charge neutrality, the substitution of PO43– by SiO44– in the hexagonal apatite lattice is accompanied by vacancy formation in the X site. Beyond the limit of y = 1–1.2, i.e. with ~50% X site emptied, the hexagonal structure becomes unstable (e.g. Gomes et al., Reference Gomes, Nedelec, Jallot and Sheptyakov2011; Marchat et al., Reference Marchat, Zymelka, Coelho, Gremillard, Joly-Pottuz, Babonneau, Esnouf, Chevalier and Bernache-Assollant2013 and references cited therein). A similar conclusion was reached in the dehydration experiments of hydroxylapatite, in which the space-group symmetry of partially dehydrated hydroxylapatite changes from hexagonal to triclinic when more than ~35% of the structurally bound water is removed (e.g. Alberius-Henning et al., Reference Alberius-Henning, Adolfsson, Grins and Fitch2001 and references cited therein). In other words, the hexagonal apatite structure could be stabilised only by stuffing > ~50% OH–, F– or Cl– into the X site. Based on these considerations, tsangpoite with the Si-enriched silicocarnotite stoichiometry, i.e. Ca10(PO4)4(SiO4)2, might be less likely to adopt the assumed apatite-like structure, despite the rather similar reflection data. However, even with the above considerations, it is still an open question as to whether apatite-structured tsangpoite with an empty c channel could be stabilised by the abundant aliovalent cations and the charge compensating vacancies, somewhat like the volume stabilisation effect in the synthetic lead compounds Pb9(PO4)6, Pb4Na(VO4)3 and Pb8K2(PO4)6 (e.g. Dicken and Brown, Reference Dickens and Brown1971; Hata et al., Reference Hata, Marumo, Iwai and Aoki1980; Mathew et al., Reference Mathew, Brown, Austin and Negas1980; White and Zhili, Reference White and Zhili2003).
On the other hand, the unique structural and chemical characteristics of tsangpoite do suggest that tsangpoite is probably the high-temperature hexagonal polymorph of silicocarnotite and has its petrogenetic origin related to the ‘continuous’ high-temperature solid-solution phase analogous to the high-temperature α-Ca2SiO4 in the binary system between α-Ca2SiO4 (hexagonal; Mumme et al., Reference Mumme, Cranswick and Chakoumakos1996) and ![]() $\bar \alpha $-Ca3(PO4)2 (trigonal; Yashima and Sakai, Reference Yashima and Sakai2003), as evidenced by the close agreement between the present electron diffraction data of tsangpoite and the XRD data of the high-T silico-phosphates stable at temperatures > ~1450°C (Tables 1 and 2). The existence of a continuous solid solution between two end-members of different stoichiometry and symmetry is somewhat unusual, but can be rationalised on a structural basis. In this regard, Bredig (Reference Bredig1942, Reference Bredig1943) suggested that the continuous solid-solution series can be considered as
$\bar \alpha $-Ca3(PO4)2 (trigonal; Yashima and Sakai, Reference Yashima and Sakai2003), as evidenced by the close agreement between the present electron diffraction data of tsangpoite and the XRD data of the high-T silico-phosphates stable at temperatures > ~1450°C (Tables 1 and 2). The existence of a continuous solid solution between two end-members of different stoichiometry and symmetry is somewhat unusual, but can be rationalised on a structural basis. In this regard, Bredig (Reference Bredig1942, Reference Bredig1943) suggested that the continuous solid-solution series can be considered as ![]() $\bar \alpha $-Ca3(PO4)2 dissolution into the hexagonal α-Ca2SiO4 lattice, forming the high-temperature hexagonal phase isotypic with α-K2SO4. Alternatively, Dickens and Brown (Reference Dickens and Brown1971) indicated that the α-Ca2SiO4, Ca5(PO4)2(SiO4), and
$\bar \alpha $-Ca3(PO4)2 dissolution into the hexagonal α-Ca2SiO4 lattice, forming the high-temperature hexagonal phase isotypic with α-K2SO4. Alternatively, Dickens and Brown (Reference Dickens and Brown1971) indicated that the α-Ca2SiO4, Ca5(PO4)2(SiO4), and ![]() $\bar \alpha $-Ca3(PO4)2 phases are all related structurally to the hexagonal K3Na(SO4)2 (aphthitalite) with certain amounts of vacant cation positions, thereby forming a continuous solid-solution series at high temperatures. The solid solution with the stoichiometry Ca5(PO4)2SiO4 is unquenchable, and transforms to orthorhombic silicocarnotite upon cooling at 1450°C, according to high-temperature camera measurements (see phase diagram in Nurse et al., Reference Nurse, Welch and Gutt1959 or Fix et al., Reference Fix, Heymann and Heinke1969). Except for the absence of reflections corresponding to the three largest d spacings of tsangpoite, the reflections from high-temperature camera powder measurements of the phase Ca5(PO4)2(SiO4) at 1550°C, as well as the reflections from the quenched high-T phases Ca11.1(PO4)1.8(SiO4)4.2 and Ca7(PO4)2(SiO4)2 (see Table 1 and phase diagram in Nurse et al., Reference Nurse, Welch and Gutt1959) are in good agreement with those of tsangpoite determined from electron diffraction. Tsangpoite is probably structurally related to the high-T silico-phosphates derived from the high-T hexagonal α-Ca2SiO4 (a = 5.53 Å, c = 7.31 Å; P63/mmc) (e.g. Yamaguchi et al., Reference Yamaguchi, Ono, Kawamura and Soda1963; Table 2), as discussed below.
$\bar \alpha $-Ca3(PO4)2 phases are all related structurally to the hexagonal K3Na(SO4)2 (aphthitalite) with certain amounts of vacant cation positions, thereby forming a continuous solid-solution series at high temperatures. The solid solution with the stoichiometry Ca5(PO4)2SiO4 is unquenchable, and transforms to orthorhombic silicocarnotite upon cooling at 1450°C, according to high-temperature camera measurements (see phase diagram in Nurse et al., Reference Nurse, Welch and Gutt1959 or Fix et al., Reference Fix, Heymann and Heinke1969). Except for the absence of reflections corresponding to the three largest d spacings of tsangpoite, the reflections from high-temperature camera powder measurements of the phase Ca5(PO4)2(SiO4) at 1550°C, as well as the reflections from the quenched high-T phases Ca11.1(PO4)1.8(SiO4)4.2 and Ca7(PO4)2(SiO4)2 (see Table 1 and phase diagram in Nurse et al., Reference Nurse, Welch and Gutt1959) are in good agreement with those of tsangpoite determined from electron diffraction. Tsangpoite is probably structurally related to the high-T silico-phosphates derived from the high-T hexagonal α-Ca2SiO4 (a = 5.53 Å, c = 7.31 Å; P63/mmc) (e.g. Yamaguchi et al., Reference Yamaguchi, Ono, Kawamura and Soda1963; Table 2), as discussed below.
Given the unit-cell correspondence a tsa ≈ <10![]() $\bar 1 $0>a; c tsa ≈ c a, tsangpoite has a triple unit-cell volume of α-Ca2SiO4 as outlined in the atomic projections along <0001> (see Fig. S5). Such an α-Ca2SiO4-derived unit cell contains 12 M sites and 6 T sites, including 2 M1 at (0, 0 and ½; site symmetry 63), 2 M2 at (⅔, ⅓, z; site symmetry 3) and (⅓, ⅔, z + ½; site symmetry 3), and 2 M3 at (⅔, ⅓, z + ½; site symmetry 3) and (⅓, ⅔, z; site symmetry 3) on three MMM… columns, as well as 6 M4 + 6 T1 (site symmetry 1) on six TMTM… columns, in accordance with the symmetry operation of the candidate P63 or P63/m space group of tsangpoite determined by electron diffraction. Given such a space group with an ideal Ca10Si2P4O24 stoichiometry, tsangpoite would have a random distribution of 2 Si and 4 P at the T1 site. The two vacant M sites per unit cell can then be assigned to 2 M1, 2 M2, or 2 M3. The first configuration by the selective vacant 2 M1 results in a fully empty cation channel along the 63 axis. This might be energetically as unstable as the empty anion channel in the non-realistic apatite-structured tsangpoite, and therefore should be discarded. On the other hand, the second configuration by the selective vacant 2 M2 or 2 M3 is energetically more favoured to have one M1–M1…, two M2–□… (or two M3–□…), and six T1–M4… columns per unit basal area for the selective P63 space group. Only when M2, M3, M4 and T1 are all at special positions, i.e. with m symmetry at z = ¼, ¾, and all TO4 tetrahedra are properly oriented, could tsangpoite have the alternative P63/m space group. Note that there is a slight deviation of M2 or M3 sites (equivalent in P63/mmc) from mirror planes in the parental α-Ca2SiO4 structure (Mumme et al., Reference Mumme, Cranswick and Chakoumakos1996). Aside from the two aforementioned ‘vacant’ M sites in the solid solution involving
$\bar 1 $0>a; c tsa ≈ c a, tsangpoite has a triple unit-cell volume of α-Ca2SiO4 as outlined in the atomic projections along <0001> (see Fig. S5). Such an α-Ca2SiO4-derived unit cell contains 12 M sites and 6 T sites, including 2 M1 at (0, 0 and ½; site symmetry 63), 2 M2 at (⅔, ⅓, z; site symmetry 3) and (⅓, ⅔, z + ½; site symmetry 3), and 2 M3 at (⅔, ⅓, z + ½; site symmetry 3) and (⅓, ⅔, z; site symmetry 3) on three MMM… columns, as well as 6 M4 + 6 T1 (site symmetry 1) on six TMTM… columns, in accordance with the symmetry operation of the candidate P63 or P63/m space group of tsangpoite determined by electron diffraction. Given such a space group with an ideal Ca10Si2P4O24 stoichiometry, tsangpoite would have a random distribution of 2 Si and 4 P at the T1 site. The two vacant M sites per unit cell can then be assigned to 2 M1, 2 M2, or 2 M3. The first configuration by the selective vacant 2 M1 results in a fully empty cation channel along the 63 axis. This might be energetically as unstable as the empty anion channel in the non-realistic apatite-structured tsangpoite, and therefore should be discarded. On the other hand, the second configuration by the selective vacant 2 M2 or 2 M3 is energetically more favoured to have one M1–M1…, two M2–□… (or two M3–□…), and six T1–M4… columns per unit basal area for the selective P63 space group. Only when M2, M3, M4 and T1 are all at special positions, i.e. with m symmetry at z = ¼, ¾, and all TO4 tetrahedra are properly oriented, could tsangpoite have the alternative P63/m space group. Note that there is a slight deviation of M2 or M3 sites (equivalent in P63/mmc) from mirror planes in the parental α-Ca2SiO4 structure (Mumme et al., Reference Mumme, Cranswick and Chakoumakos1996). Aside from the two aforementioned ‘vacant’ M sites in the solid solution involving ![]() $\bar \alpha $-Ca3(PO4)2 dissolution into the hexagonal α-Ca2SiO4 lattice (Bredig, Reference Bredig1942, Reference Bredig1943), there are additional 0.84 M vacancies per tsangpoite unit cell for charge compensation due to the substitution of aliovalent cations Ti4+, Fe3+, Al3+, Y6+ and REE 6+ for Ca2+, which are therefore most probably randomly distributed throughout the tsangpoite structure.
$\bar \alpha $-Ca3(PO4)2 dissolution into the hexagonal α-Ca2SiO4 lattice (Bredig, Reference Bredig1942, Reference Bredig1943), there are additional 0.84 M vacancies per tsangpoite unit cell for charge compensation due to the substitution of aliovalent cations Ti4+, Fe3+, Al3+, Y6+ and REE 6+ for Ca2+, which are therefore most probably randomly distributed throughout the tsangpoite structure.
The α-Ca2SiO4-derived superlattice and the cation/vacancy ordering scheme are also of concern to the structural similarity/difference between nagelschmidtite, silicocarnotite and tsangpoite. Nagelschmidtite Ca7□(PO4)2(SiO4)2 with the assigned P61 space group and unit-cell correspondence a nag ≈ 2a a, c nag ≈ c a (where a nag = 10.78 Å, c nag =21.42 Å, P6 1 and Z = 6) (see Fig. S5) has one MMM… chain (61 axis), three equivalent MMM… chains (21 axis), six equivalent TMTM… chains (31 axis), and other two equivalent TMTM… chains (Widmer et al., Reference Widmer, Gfeller and Armbruster2015). Given the M 42T 24O96 stoichiometry deviating from 48 M sites and 24 T sites in the unit cell, nagelschmidtite requires six vacant M sites at different heights of the individual six equivalent TMTM… chains to yield six T□TMTMT□T… chains in accordance with the 61 and 31 symmetry of the P61 space group (see projection in fig. 8 of Widmer et al., Reference Widmer, Gfeller and Armbruster2015). Furthermore, following the screw axis 61 rather than 63 of tsangpoite, nagelschmidtite is expected to have a c axis unit repetition distance three times that of tsangpoite when derived from α-Ca2SiO4. This is indeed the case with c = ~21 Å for nagelschmidtite and c = ~7 Å for tsangpoite. Although with much more zigzag MMM… or TMTM…. chains along the a axis, silicocarnotite Ca10□(PO4)4(SiO4)2 can be visualised as the structural derivative of α-Ca2SiO4 by the unit-cell correspondence a sca ≈ c a, b sca ≈ 3a a, c sca ≈ √3c a (where a sca = 6.72 Å, b sca = 15.45 Å and c sca =10.08 Å, Pnma and Z = 2) (see Fig. S5), or be seen as a 1:1 alternation of stretched α’(H)-Ca2SiO4 modules (Si/P = 1), intersliced by Ca(PO4)– modules (Galuskin et al., Reference Galuskin, Galuskina, Gfeller, Krüger, Kusz, Vapnik, Dulski and Dzierżanowski2016). Given that the M 20T 12O48 stoichiometry deviates from the allowed 24 M sites and 12 T sites, silicocarnotite requires four vacant M sites in the unit cell. As there are six general equivalent positions for the Pnma space group, the four vacancies can only be placed along either two equivalent MM… chains or four equivalent TMTM… chains sitting on the {010} mirror planes (y = ¼ and ¾) (e.g. see projection in fig. 11 of Galuskin et al., Reference Galuskin, Galuskina, Gfeller, Krüger, Kusz, Vapnik, Dulski and Dzierżanowski2016). Similar to that noted in tsangpoite, the first configuration would yield an unstable configuration with two fully empty cation chains along the a axis, and hence should be discarded. Instead, the four vacancies have to be placed on the four individual equivalent TMTM… chains on the {010} mirrors, thereby yielding four T□T□T… chains along the a axis (see structural refinement in Galuskin et al., Reference Galuskin, Galuskina, Gfeller, Krüger, Kusz, Vapnik, Dulski and Dzierżanowski2016). In contrast to the above cases with vacancies at the specific M sites, the vacancies in the synthetic flamite-like phase Ca15□(PO4)2(SiO4)6 by the unit-cell correspondence a fla ≈ √3c a, b fla ≈ 4a a, c fla ≈ c a (where a fla = 9.40 Å, b fla = 21.71 Å, c fla =6.83 Å, Pnm21 and Z = 2) were considered most likely to be placed randomly at M2 and M3 along TMTM…columns, such as Na in flamite Ca14Na2 (PO4)2(SiO4)6 (Saalfeld and Klaska, Reference Saalfeld and Klaska1981, Gfeller et al., Reference Gfeller, Widmer, Krüger, Galuskin and Armbruster2015; Widmer et al., Reference Widmer, Gfeller and Armbruster2015).
In contrast to the cation–vacancy ordered structures of tsangpoite and nagelschmidtite, the high-T silico-phosphates in the α-Ca2SiO4–![]() $\bar \alpha $-Ca3(PO4)2 system most likely have the high-temperature disordered structure with all the vacancies distributed randomly in M sites to give the common α-Ca2SiO4-like unit cell with fewer reflections than tsangpoite (see Table 1). Except for the composition range ~25–40 wt.% Ca3(PO4)2 having the high-T phase stability field extending down to ~<1200°C for sluggish transformation kinetics, the high-T silico-phosphates in the Ca2SiO4–Ca3(PO4)2 system were known to be unquenchable (Nurse et al., Reference Nurse, Welch and Gutt1959; Fix et al., Reference Fix, Heymann and Heinke1969) due to high equilibrium temperatures for rather rapid phase transformation upon cooling/quenching. However, it was noted the high-T phase stability field of the eutectoid composition with 35 wt.% Ca3(PO4)2 (at ~500°C) may well extend to room temperature due to the impurities (Nurse et al., Reference Nurse, Welch and Gutt1959). The impurity stabilisation effect on the high-T phase was also noted in the Na2O, Fe2O3 and/or Al2O3 stabilised α-Ca2SiO4 solid-solution phases, which were in fact quenchable from T > 1400°C (Bredig, Reference Bredig1943; Fukuda et al., Reference Fukuda, Maki, Toyoda and Ito1993; see Table 2) and yet the pure α-Ca2SiO4 was unquenchable (Nurse et al., Reference Nurse, Welch and Gutt1959; Fix et al., Reference Fix, Heymann and Heinke1969). The time-temperature-transformation diagram of α to α’H transition of Ca2SiO4 solid solution with different concentrations of foreign oxides (Na2O, Fe2O3, and Al2O3) further showed that the kinetic cut-off temperature and the activation energy for the growth of α’H increase steadily with increasing concentration of impurities (Fukuda et al., Reference Fukuda, Maki, Toyoda and Ito1993). It is thus possible that a high content of Ti4+, Fe3+ and Al3+, as tramp impurities, may extend the high-T-phase stability field to lower temperatures, around 1200°C, to allow vacancy ordering for tsangpoite formation, and also to avoid the high-T phase → silicocarnotite transformation upon cooling/quenching as reported here.
$\bar \alpha $-Ca3(PO4)2 system most likely have the high-temperature disordered structure with all the vacancies distributed randomly in M sites to give the common α-Ca2SiO4-like unit cell with fewer reflections than tsangpoite (see Table 1). Except for the composition range ~25–40 wt.% Ca3(PO4)2 having the high-T phase stability field extending down to ~<1200°C for sluggish transformation kinetics, the high-T silico-phosphates in the Ca2SiO4–Ca3(PO4)2 system were known to be unquenchable (Nurse et al., Reference Nurse, Welch and Gutt1959; Fix et al., Reference Fix, Heymann and Heinke1969) due to high equilibrium temperatures for rather rapid phase transformation upon cooling/quenching. However, it was noted the high-T phase stability field of the eutectoid composition with 35 wt.% Ca3(PO4)2 (at ~500°C) may well extend to room temperature due to the impurities (Nurse et al., Reference Nurse, Welch and Gutt1959). The impurity stabilisation effect on the high-T phase was also noted in the Na2O, Fe2O3 and/or Al2O3 stabilised α-Ca2SiO4 solid-solution phases, which were in fact quenchable from T > 1400°C (Bredig, Reference Bredig1943; Fukuda et al., Reference Fukuda, Maki, Toyoda and Ito1993; see Table 2) and yet the pure α-Ca2SiO4 was unquenchable (Nurse et al., Reference Nurse, Welch and Gutt1959; Fix et al., Reference Fix, Heymann and Heinke1969). The time-temperature-transformation diagram of α to α’H transition of Ca2SiO4 solid solution with different concentrations of foreign oxides (Na2O, Fe2O3, and Al2O3) further showed that the kinetic cut-off temperature and the activation energy for the growth of α’H increase steadily with increasing concentration of impurities (Fukuda et al., Reference Fukuda, Maki, Toyoda and Ito1993). It is thus possible that a high content of Ti4+, Fe3+ and Al3+, as tramp impurities, may extend the high-T-phase stability field to lower temperatures, around 1200°C, to allow vacancy ordering for tsangpoite formation, and also to avoid the high-T phase → silicocarnotite transformation upon cooling/quenching as reported here.
Lastly, it is emphasised that, based on the present work, tsangpoite indeed has a hexagonal crystal structure (P63 or P63/m) with a unit cell similar to apatite or the high-temperature hexagonal phase in the Ca2SiO4–Ca3(PO4)2 system. Its crystal structure along with chemical composition is distinctly different from existing known mineral phases, and tsangpoite is accordingly a distinct mineral species in nature. Unfortunately, the present electron diffraction data are unable to distinguish if the tsangpoite crystal structure is more similar to an apatite structure with empty channels or to the α-Ca2SiO4-derived structure, nor do they clearly define the possible presence of small cation sites associated with Ti and/or Fe and the abundant structure vacancies as indicated in the empirical formula. Future single-crystal XRD studies on larger tsangpoite crystals than the tiny ones in the present study by four cycle diffractometers, coupled with further detailed spectroscopic evidence, may solve this problem.
Matyhite
Merrillite, also known as ‘whitlockite’ in the early literature, is one of the main Ca phosphate minerals, along with apatite, that occur mainly in Martian meteorites and Lunar rocks, and rarely in terrestrial pyrometamorphic rocks (e.g. Galuskina et al., Reference Galuskina, Galuskin and Vapnik2016). Significant structural differences between terrestrial whitlockite Ca9Mg(PO4)6(PO3OH) and the extra-terrestrial merrillite Ca9NaMg(PO4)7, e.g. the split of the P1 site in merrillite into P(A) and P(A’) sites and the consequent inversion of the P(A) tetrahedron with incorporation of hydrogen atoms in whitlockite (e.g. Gopal and Calvo, Reference Gopal and Calvo1972; Dowty, Reference Dowty1977; Yashima et al., Reference Yashima, Sakai, Kamiyama and Hoshikawa2003; Hughes et al., Reference Hughes, Jolliff and Gunter2006, Jolliff et al., Reference Jolliff, Hughes, Freeman and Zeigler2006; Hughes et al., Reference Hughes, Jolliff and Rakovan2008), suggest that they are in fact two different minerals and the exclusive usage of ‘merrillite’ for the hydrogen-free extra-terrestrial varieties is warranted (Dowty, Reference Dowty1977; Rubin, Reference Rubin1997). Not considering the limited substitution of Si for P at tetrahedral sites, the compositions of extra-terrestrial merrillite varieties can be described by the general formula (Ca9–x–z[Y,REE]x+z)Σ9Na1–x(Mg,Fe,Mn)Σ1(P7–zSiz)Σ7O28, and can be plotted in a ternary diagram: Ca-merrillite Ca9(Ca0.5□0.5)(Mg,Fe,Mn)Σ1(PO4)7, Na-merrillite Ca9Na(Mg,Fe,Mn)Σ1(PO4)7 [i.e. ‘merrillite’, approved by the International Mineralogical Association (IMA) in 1977] and REE-merrillite (Ca8[Y,REE]1)□(Mg,Fe,Mn)Σ1(PO4)7 (see Fig. 10 modified from fig. 7 in Shearer et al., Reference Shearer, Burger, Papike, McCubbin and Bell2015) (e.g. Dowty, Reference Dowty1977; Jolliff et al., Reference Jolliff, Hughes, Freeman and Zeigler2006; Shearer et al., Reference Shearer, Burger, Papike, McCubbin and Bell2015). As ferromerrillite Ca9NaFe(PO4)7 from the Shergotty meteorite has been considered as the Fe-analogue of Na-merrillite Ca9NaMg(PO4)7 and approved as a new merrillite-group mineral by the IMA (Britvin et al., Reference Britvin, Krivovichev and Armbruster2016; IMA2006-039), the present matyhite Ca9(Ca0.5□0.5)Fe(PO4)7 with the Na site (irregular 6-fold coordinated in merrillite structure) half empty and half occupied by Ca and the Mg site (regular 6-fold coordinated in merrillite structure) fully occupied by Fe, is in fact the Fe-analogue of Ca-merrillite Ca9(Ca0.5□0.5)Mg(PO4)7 and should therefore be considered as a new species in the merrillite group, following the Criteria for New Mineral Species as detailed in Nickel and Grice (Reference Nickel and Grice1998).
Fig. 10. Ternary diagram illustrating the composition variations of extra-terrestrial merrillites versus Ca-merrillite, matyhite, Na-merrillite, ferromerrillite, and REE-merrillite (modified from fig. 7 in Shearer et al., Reference Shearer, Burger, Papike, McCubbin and Bell2015).
The chemical variations among merrillites in Martian meteorites, Lunar rocks, and the D'Orbigny angrite can be illustrated using the Ca-merrillite, Na-merrillite and REE-merrillite ternary diagram (see Fig. 10 modified from fig. 7 in Shearer et al., Reference Shearer, Burger, Papike, McCubbin and Bell2015). Due to low abundance in REE, the Martian merrillites mostly lie along the Ca-merrillite–Na-merrillite join, and are dominated by the coupled substitution CaNa-site + □Na-site ⇔ 2NaNa-site. In contrast to the Martian merrillites, Lunar merrillites have a large REE-merrillite component, and are therefore displaced from the Ca-merrillite–Na-merrillite join towards the REE-merrillite apex. Among all extra-terrestrial merrillites, matyhite has the greatest Ca, Fe and Si abundances, i.e. 9.33 Ca atoms, 0.94 Fe atoms and 0.26 Si atoms per 28 oxygen atoms (Table 8), and appears to be a rather rare mineral species in terms of composition. Among the ample chemical data of Fe-rich merrillite minerals from Martian meteorites in Shearer et al. (Reference Shearer, Burger, Papike, McCubbin and Bell2015), there are only two analyses having similar Na-site occupancies with Ca > Na + K, comparable to matyhite (see Table 8). As for the occasional merrillite minerals from plutonic angrites, their Mg-, Ca- and Na-site occupancies are closer to Ca-merrillite (Prinz et al., Reference Prinz, Keil, Hlava, Berkley, Gomes and Curvello1977; McKay et al., Reference McKay, Lindstrom, Yang and Wagstaff1988) (see Table 8).
The formation of tsangpoite, matyhite and kuratite in the D'Orbigny angrite
Aside from the debate on the possible non-igneous origin of the D'Orbigny angrite (Mittlefehldt et al., Reference Mittlefehldt, Killgore and Lee2002; Kurat et al., Reference Kurat, Varela, Brandstätter, Weckwerth, Clayton, Weber, Schultz, Wäsch and Nazarov2004; Varela et al., Reference Varela, Kurat, Zinner, Hoppe, Ntaflos and Nazarov2005; Reference Varela, Hwang, Shen, Chu, Yui, Iizuka, Brandstätter and Abdu2017), angrites are usually considered to be basaltic igneous rocks. Following the igneous scenario, the accessory minerals kuratite, matyhite and tsangpoite, along with Al–Ti-bearing hedenbergite, troilite, and Ca and Fe-rich olivine, are considered as mesostasis phases crystallised from residue magmas (e.g. Mittlefehldt et al., Reference Mittlefehldt, Killgore and Lee2002; Keil, Reference Keil2012), similar to the sequential crystallisation of merrillite and apatite from magma in Martian meteorites and/or Lunar rocks (e.g. Greenwood et al., Reference Greenwood, Blake and Coath2003; Gross et al., Reference Gross, Filiberto, Herd, Melwani Daswani, Schwenzer and Treiman2013), or the formation of Fe2+-dominant rhönites in terrestrial undersaturated basaltic rocks (Gamble and Kyle, Reference Gamble and Kyle1987; Grapes and Keller, Reference Grapes and Keller2010; Havette et al., Reference Havette, Clocchiatti, Nativel and Montaggioni1982; Olsson, Reference Olsson1983). In accordance with the above, the occurrence of kuratite, matyhite and tsangpoite embedded in hedenbergite or the Ca-rich fayalite + kirschsteinite intergrowth rim of resorbed Fe sulfide suggests that these phases could have crystallised from an interstitial melt at a very late stage of rock formation. This interstitial melt must be subsilicic, almost Mg-, Na- and K-free but enriched in Ca, Fe, Ti, Al and P.
Alternatively, the close petrographic relations between tsangpoite/matyhite and the resorbed Fe sulfide rimed by the Fa + Kir symplectite, such as: (1) the nucleation of tsangpoite with magnetite ± other phases as inclusions in the local partial melting-like domain in ‘intact’ Fe sulfide (Fig. 2a–c) or at the core of resorbed Fe sulfide (Fig. 5a); (2) the elongated tsangpoite crystal extending outward from the core of an Fe sulfide grain into hedenbergite (Figs 3f, 5c); (3) the commonly observed bundles of sub-parallel, elongated tsangpoite crystals growing from the Fa + Kir symplectic rim of Fe sulfide outward into hedenbergite (see Figs 1a,b, 3b,c); and (4) sets of dendritic matyhite plates subparallel within the Fa + Kir symplectic rim of Fe sulfide extending outward into hedenbergite (Figs 1d, 4d,e); would infer that these new minerals probably represent metasomatic products resulting from reactions between an intruding Ca–Ti–Al–P–O-rich metasomatic agent, which could be a secondary melt (as suggested in Mittlefehldt et al., Reference Mittlefehldt, Killgore and Lee2002), a liquid or gas, and the porous olivine–plagioclase plate + Fa–Kir overgrowth (over olivine) + augite + Fe sulfide aggregates, similar to the formation process proposed for kuratite (Hwang et al., Reference Hwang, Shen, Chu, Yui, Varela and Iizuka2016a) and in accordance with the non-igneous genetic processes of the D'Orbigny angrite proposed by Kurat et al. (Reference Kurat, Varela, Brandstätter, Weckwerth, Clayton, Weber, Schultz, Wäsch and Nazarov2004) and Varela et al. (Reference Varela, Kurat, Zinner, Métrich, Brandstätter, Ntaflos and Sylvester2003). In either case, the formation sequence is probably tsangpoite → matyhite → kuratite → Fa + Kir symplectite (over resorbed Fe sulfide) → hedenbergite, according to petrogenetic relationships, and the crystallisation temperatures must be >1000°C, considering the: co-presence of Ca-rich fayalite and kirschsteinite (Mukhopadhyay and Lindsley, Reference Mukhopadhyay and Lindsley1983; Sokol et al., Reference Sokol, Sharygin, Kalugin, Volkova and Nigmatulina2002); the stabilisation of the ordered tsangpoite structure by Fe2O3 and Al2O3 at ~1200°C (this study); the partial melting of Fe sulfide at ~1200°C (Ehfers, Reference Ehfers1972) as evidenced by the FeS droplets in matyhite plates (Fig. 4c–f); and the 950–1180°C stability field of rhönites based on the available experimental (Kunzmann, Reference Kunzmann1999) and empirical (e.g. Grapes and Keller, Reference Grapes and Keller2010; Sharygin et al., Reference Sharygin, Kthay, Szab, Timina, Trk, Vapnik and Kuzmin2011; Peretyazhko et al., Reference Peretyazhko, Savina and Khromova2017) observations.
Whereas the above scenarios rationalise the occurrences of euhedral tsangpoite, matyhite and kuratite in D'Orbigny, some issues regarding the formation mechanisms/conditions of these new minerals require further consideration/investigation. First of all, unless it was due to the extreme Fe3+ partition in tsangpoite involving unknown kinetics problems, the very high Fe3+/ΣFe ratio (~1) of tsangpoite (this study; Mikouchi et al., Reference Mikouchi, Sugiyama, Satake and Amelin2011), compared to the values of ~0.3, ~0.05 and ~0.15, respectively, for matyhite (this study), kuratite (Hwang et al., Reference Hwang, Shen, Chu, Yui, Varela and Iizuka2016a) and ulvöspinel (see Table S1b from Mittlefehldt et al., Reference Mittlefehldt, Killgore and Lee2002), might reflect the change in ![]() $f_{{\rm O}_{\rm 2}}$ during crystallisation of grain-boundary phases in D'Orbigny. However, without proper temperature constraints, it is hard to address this issue further. Besides, unlike the common merrillite–apatite intergrowths due to sequential crystallisation in Martian magma (e.g. Greenwood et al., Reference Greenwood, Blake and Coath2003; Gross et al., Reference Gross, Filiberto, Herd, Melwani Daswani, Schwenzer and Treiman2013), tsangpoite and matyhite occur exclusively in different hedenbergite domains at the contact with Fa–Kir intergrowths. Unless by melt immiscibility similar to that in calc-alkali phosphate–silicate systems (e.g. Toropov et al., Reference Toropov, Barzakovskiv, Lapin, Kurtseva and Baykova1972; Rabinovich et al., Reference Rabinovich, Ish-Shalom and Kisilev1980) or some unknown causes, it is hard to envision how the composition of a melt phase could vary so greatly that two phosphate minerals of different compositions and structures would separately crystallise at different domains in the residue melt. Further study is also required to clarify how a crystallisation/growth process could possibly yield the elongated tsangpoite crystal with the characteristic, continuous central microtube filled by magnetite + ferrite + Fe–S–O (Figs 1c, 3e,f), and with the pronounced bimodal crystal sizes as illustrated in Fig. 2d–i.
$f_{{\rm O}_{\rm 2}}$ during crystallisation of grain-boundary phases in D'Orbigny. However, without proper temperature constraints, it is hard to address this issue further. Besides, unlike the common merrillite–apatite intergrowths due to sequential crystallisation in Martian magma (e.g. Greenwood et al., Reference Greenwood, Blake and Coath2003; Gross et al., Reference Gross, Filiberto, Herd, Melwani Daswani, Schwenzer and Treiman2013), tsangpoite and matyhite occur exclusively in different hedenbergite domains at the contact with Fa–Kir intergrowths. Unless by melt immiscibility similar to that in calc-alkali phosphate–silicate systems (e.g. Toropov et al., Reference Toropov, Barzakovskiv, Lapin, Kurtseva and Baykova1972; Rabinovich et al., Reference Rabinovich, Ish-Shalom and Kisilev1980) or some unknown causes, it is hard to envision how the composition of a melt phase could vary so greatly that two phosphate minerals of different compositions and structures would separately crystallise at different domains in the residue melt. Further study is also required to clarify how a crystallisation/growth process could possibly yield the elongated tsangpoite crystal with the characteristic, continuous central microtube filled by magnetite + ferrite + Fe–S–O (Figs 1c, 3e,f), and with the pronounced bimodal crystal sizes as illustrated in Fig. 2d–i.
Concluding remarks
Two new minerals: tsangpoite (silicate–phosphate) and matyhite (phosphate), mainly in association with kuratite, ulvöspinel, hedenbergite, Ca and Fe olivine, and Fe sulfide were identified in the D'Orbigny angrite meteorite, a possible analogue of terrestrial dolerite/diabase in texture. The kuratite and matyhite, along with kuratite, occurred as idiomorphic crystals in the marginal part of hedenbergite or in the Fa–Kir symplectite associated with Fe sulfide indicating they were probably crystallised from a residue melt. Alternatively, texture observations, especially petrographic relationships between the new minerals, Fe sulfide and Fa–Kir symplectite, point to a possible origin of these minerals from metasomatic reactions between an intruding Ca–Fe–Ti–Al–P–O-rich agent and the porous olivine-plagioclase plate + Fa–Kir overgrowth + augite + Fe sulfide aggregates, similar to the formation process of kuratite proposed by Hwang et al. (Reference Hwang, Shen, Chu, Yui, Varela and Iizuka2016a), or the genetic processes of D'Orbigny proposed by Kurat et al. (Reference Kurat, Varela, Brandstätter, Weckwerth, Clayton, Weber, Schultz, Wäsch and Nazarov2004) and Varela et al. (Reference Varela, Kurat, Zinner, Métrich, Brandstätter, Ntaflos and Sylvester2003). The unique compositions of these new minerals truly reflect the high Ca, Fe, Ti and Al, as well as the low Mg, K and Na chemical characteristics of the angrite parent body.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1180/mgm.2018.125
Acknowledgements
This research is supported by funding from MOST, Taiwan, Republic of China. Helpful suggestions and comments by reviewers are highly appreciated. We thank in particular E.V. Galuskin for pointing out the structure ambiguity of tsangpoite to be solved in future work.