Introduction
Most fungi forming symbioses with cyanobacteria (‘cyanolichens’) are currently classified in the order Peltigerales (Lecanoromycetes), a group comprising a number of large and conspicuous macrolichen families, including the Pannariaceae (Tehler & Wedin Reference Tehler, Wedin and Nash2008; Lumbsch & Huhndorf Reference Lumbsch and Huhndorf2010). The Pannariaceae at present contains c. 20 genera (Jørgensen Reference Jørgensen2003, Reference Jørgensen2008; Passo et al. Reference Passo, Stenroos and Calvelo2008), two of which are Fuscopannaria and Vahliella.
Fuscopannaria leucophaea (Vahl) P. M. Jørg. and related species were recognized as a distinct subgenus, Fuscopannaria subgenus Micropannaria, by Jørgensen (Reference Jørgensen1994). The members differ from the subgenus Fuscopannaria [type F. leucosticta (Tuck.) P. M. Jørg.] mainly in ascus characteristics, having an apical amyloid internal sheet instead of an amyloid tube structure, but also by lacking secondary compounds by TLC, by lacking a distinct epispore, and by having irregularly developed thalline ascoma margins. In several recent phylogenetic investigations of cyanobacterial lichens, samples of Fuscopannaria leucophaea appear not only outside Fuscopannaria in the strict sense but also outside a monophyletic Pannariaceae (Ekman & Jørgensen Reference Ekman and Jørgensen2002; Wedin & Wiklund Reference Wedin and Wiklund2004; Wedin et al. Reference Wedin, Jørgensen and Wiklund2007). This led Jørgensen (Reference Jørgensen2008) to describe the new genus Vahliella for Fuscopannaria subgenus Micropannaria, including eight very closely related species. Jørgensen (Reference Jørgensen2008) hesitated to exclude Vahliella from Pannariaceae until further studies had confirmed this placement. A much enlarged study of the Peltigerales based on three molecular markers and covering many more taxa (Wedin et al. Reference Wedin, Wiklund, Jørgensen and Ekman2009) resulted in a similar topology as previous studies regarding the placement of Vahliella, but did not add further Vahliella samples. Wedin et al. (Reference Wedin, Wiklund, Jørgensen and Ekman2009) concluded that additional samples and species of Vahliella should be included in future analyses to test this relationship further. They also concluded that if such investigations confirmed that Vahliella was the sister group to the rest of Peltigerales suborder Peltigerineae sensu Miądlikowska & Lutzoni (Reference Miądlikowska and Lutzoni2004), then a new family needed to be described. A study including another F. leucophaea sample (Högnabba et al. Reference Högnabba, Stenroos, Thell and Myllys2009) supported the conclusion that Vahliella did not belong to the Pannariaceae implicitly, but the authors did not discuss it. Here, we provide an extended study of this problem. We have increased the sampling of taxa in Vahliella, and test the placement of the genus in the Peltigerales to see if a new name at family level is needed as indicated by the placement in earlier investigations.
Material and Methods
DNA extractions, amplification, and sequencing
DNA extractions, amplification and sequencing of new sequences follow the methods and settings described in Wedin et al. (Reference Wedin, Wiklund, Jørgensen and Ekman2009). The markers selected were the mitochondrial SSU rDNA (mtSSU rDNA), and the gene coding for the largest subunit of RNA polymerase II (RPB1). These markers have proved useful in resolving phylogenetic relationships in the group, and to produce congruent results, in our earlier studies of cyanolichens in the Peltigerales.
Sequence alignment and phylogenetic analyses
New protein coding RPB1 sequences were unambiguously aligned to the alignment used by Wedin et al. (Reference Wedin, Wiklund, Jørgensen and Ekman2009). This alignment contained very few gaps (four internal amino acid alignment sites) and most of the variation was found to be synonymous. mtSSU rDNA sequences were aligned using the Q-INS-i algorithm (Katoh & Toh Reference Katoh and Toh2008a) of the multiple alignment software MAFFT version 6.611 (Katoh et al. Reference Katoh, Misawa, Kuma and Miyata2002; Katoh & Toh Reference Katoh and Toh2008b). The gap opening cost was set to 1·53 and the offset to 0 (i.e., default parameters). Ambiguous alignment in the mtSSU rDNA was identified and removed using Gblocks version 0.91b (Castresana Reference Castresana2000) with the relaxed condition parameters suggested by Talavera & Castresana (Reference Talavera and Castresana2007).
Phylogenetic analyses
To estimate dataset incongruence between the single-gene matrices the markers were analyzed separately by maximum parsimony bootstrapping and compared. Incongruence was defined as high bootstrap support (≥ 70 %; Hillis & Bull Reference Hillis and Bull1993) for incompatible associations (i.e., groups in conflict with each other between the single-gene analyses). No incongruence was identified and we consequently proceeded with a phylogenetic analysis of the concatenated data.
We used Bayesian inference of phylogeny as implemented in the software BayesPhylogenies 1.1 (Pagel & Meade Reference Pagel and Meade2004). We used a combination of a pattern heterogeneity mixture model (Pagel & Meade Reference Pagel and Meade2004, Reference Pagel, Meade and Gascuel2005) and a branch-length set mixture model (Meade & Pagel Reference Meade, Pagel and Pontarotti2008; Pagel & Meade Reference Pagel and Meade2008) in order to test if the phylogenetic position of Vahliella reported by Wedin et al. (Reference Wedin, Wiklund, Jørgensen and Ekman2009) could be an artefact from improper data partitioning or unaccounted for by heterotachy (rate variation across a tree). For both types of model, we used reversible-jump Markov chain Monte Carlo (MCMC) to reduce parameter redundancy and to allow a priori ignorance about the number of substitution rate matrices, the specific partitioning of the data, as well as the number of branch length sets. We allowed an unspecified number of independently parameterized general time-reversible (GTR) rate matrices coupled with gamma (Γ) distributed rate heterogeneity across sites modelled as four discrete categories. The maximum number of branch length sets allowed was two. BayesPhylogenies operates with fixed priors that cannot be modified by the user. Priors were accounted for by Pagel & Meade (Reference Pagel and Meade2008), except that the prior on branch lengths in the publicly available version of BayesPhylogenies 1.1 is exponentially distributed with mean 1 (A. Meade, personal communication). We performed six identical MCMC runs without Metropolis-coupling. Each run was 20 million generations in length, and trees and parameters were sampled every 1000 generations. The first half of each converged run was removed as burn-in. The marginal likelihood of the data was calculated using importance sampling (Newton & Raftery Reference Newton and Raftery1994; Suchard et al. Reference Suchard, Kitchen, Sinsheimer and Weiss2003) with 10 000 bootstrap replicates, as implemented in Tracer 1.4 (Rambaut & Drummond Reference Rambaut and Drummond2007).
We further used maximum parsimony and parsimony bootstrap, performed using PAUP* 4.0b10 (Swofford Reference Swofford2002). Heuristic search settings: gaps treated as ‘missing’, 1000 random addition sequence replicates, TBR branch swap, steepest descent off, collapse branches if minimum length is 0, MulTrees on. Parsimony bootstrap: bootstrap settings: 1000 bootstrap replicates, full heuristic search, retain groups with frequency >50%; heuristic search settings: as above, but 10 random addition replicates. Uninformative characters were excluded from the analyses.
Results
Taxon sampling and DNA sequencing
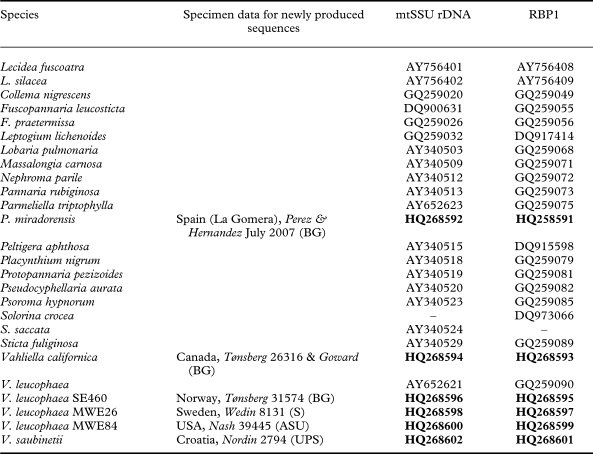
We sampled taxa representing most families currently belonging in Peltigerales (Collemataceae, Lobariaceae, Massalongiaceae, Nephromataceae, Pannariaceae, Peltigeraceae, and Placynthiaceae). We obtained 13 new mtSSU rDNA and 13 RPB1 sequences (Table 1). The genus Solorina (Peltigeraceae) was represented by an mtSSU rDNA sequence of Solorina saccata (L.) Ach. and an RPB1 sequence of S. crocea (L.) Ach. The sequences were aligned together with sequences from our earlier studies, deposited in GenBank. Lecidea fuscoatra (L.) Ach. functioned as outgroup to root the tree.
Table 1. Specimens and sequences included in this study (newly produced sequences in bold)

Phylogenetic analyses
We compiled matrices including 25 taxa. The MAFFT alignment of mtSSU rDNA was 1100 nucleotides of which 390 were suggested for removal by Gblocks. RPB1 had no ambiguous sites. The concatenated matrix included 1432 characters after removal of ambiguous sites (RPB1 722 and mtSSU rDNA 710 nucleotides) of which 670 were variable and 502 parsimony-informative. Among the 383 variable characters in the RPB1 partition, 328 were parsimony-informative, and 222 out of 287 variable characters in the mtSSU rDNA were parsimony-informative.
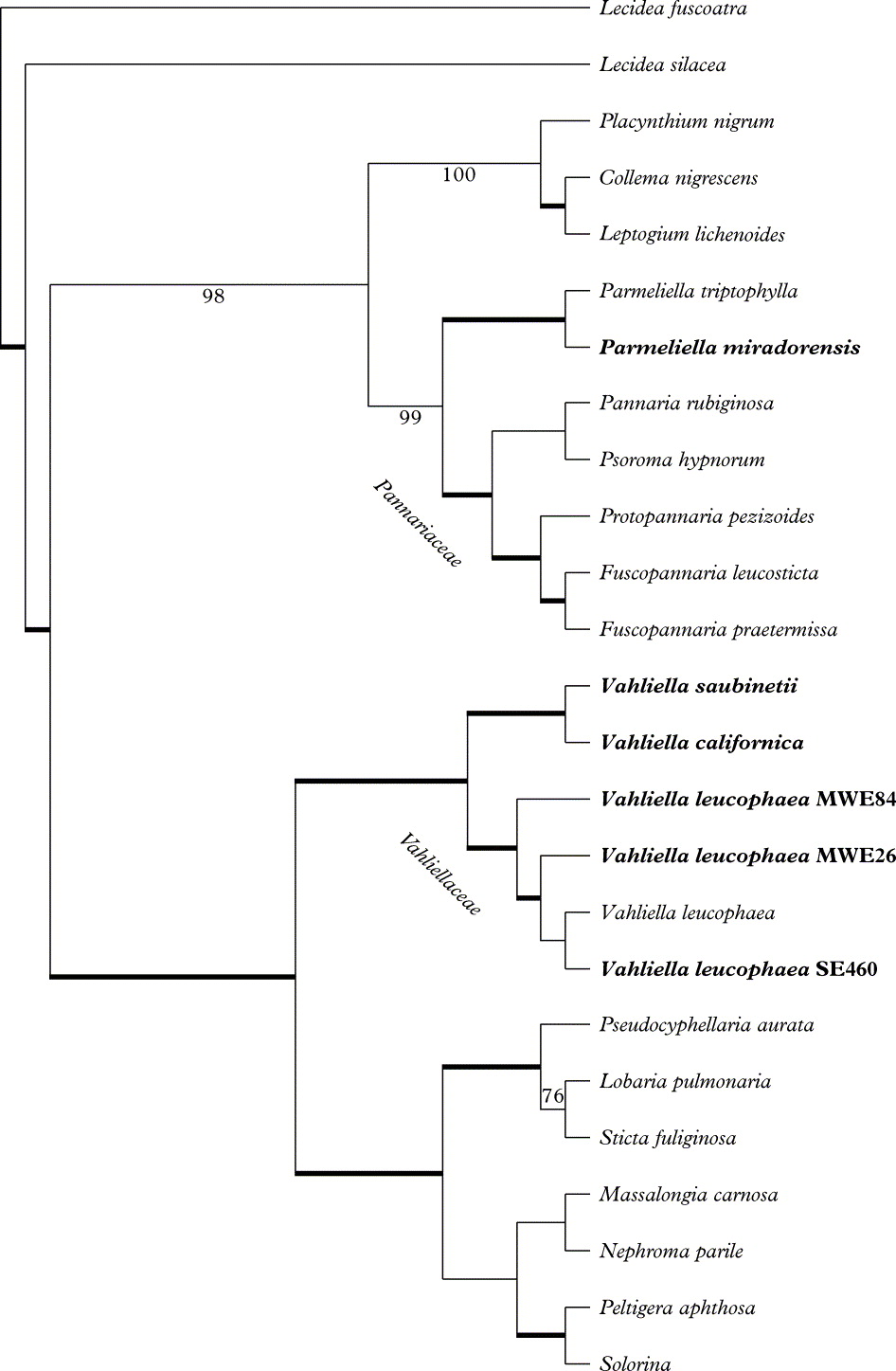
In the Bayesian analyses, three out of six MCMC runs were found to suffer from poor mixing (as revealed by low effective sample sizes) or convergence problems (as revealed by biologically unrealistic parameter values), and were consequently excluded from further analysis. The three other MCMC runs were found to display excellent mixing properties and seemed to have converged on the same likelihood and parameter values. The samples drawn from these three runs were pooled and used for further analyses. Two independent GTR+dΓ4 models were fitted to the data with 100 % probability. The 95 % highest posterior density (a Bayesian equivalent of the most frequent confidence interval) of the number of branches requiring a second branch length ranged from zero to ten, the median being 3·9. No individual branch in the MCMC tree sample had a probability exceeding 50% of having a second length. The majority-rule consensus tree based on 30 000 trees is presented in Fig. 1. The MP analyses resulted in 6 most parsimonious trees of 1927 steps, CI = 43 and RI = 59. Groupings significantly supported in both analyses (≥95 Bayesian PP and ≥70% MP-BS) are indicated with thick internodes.
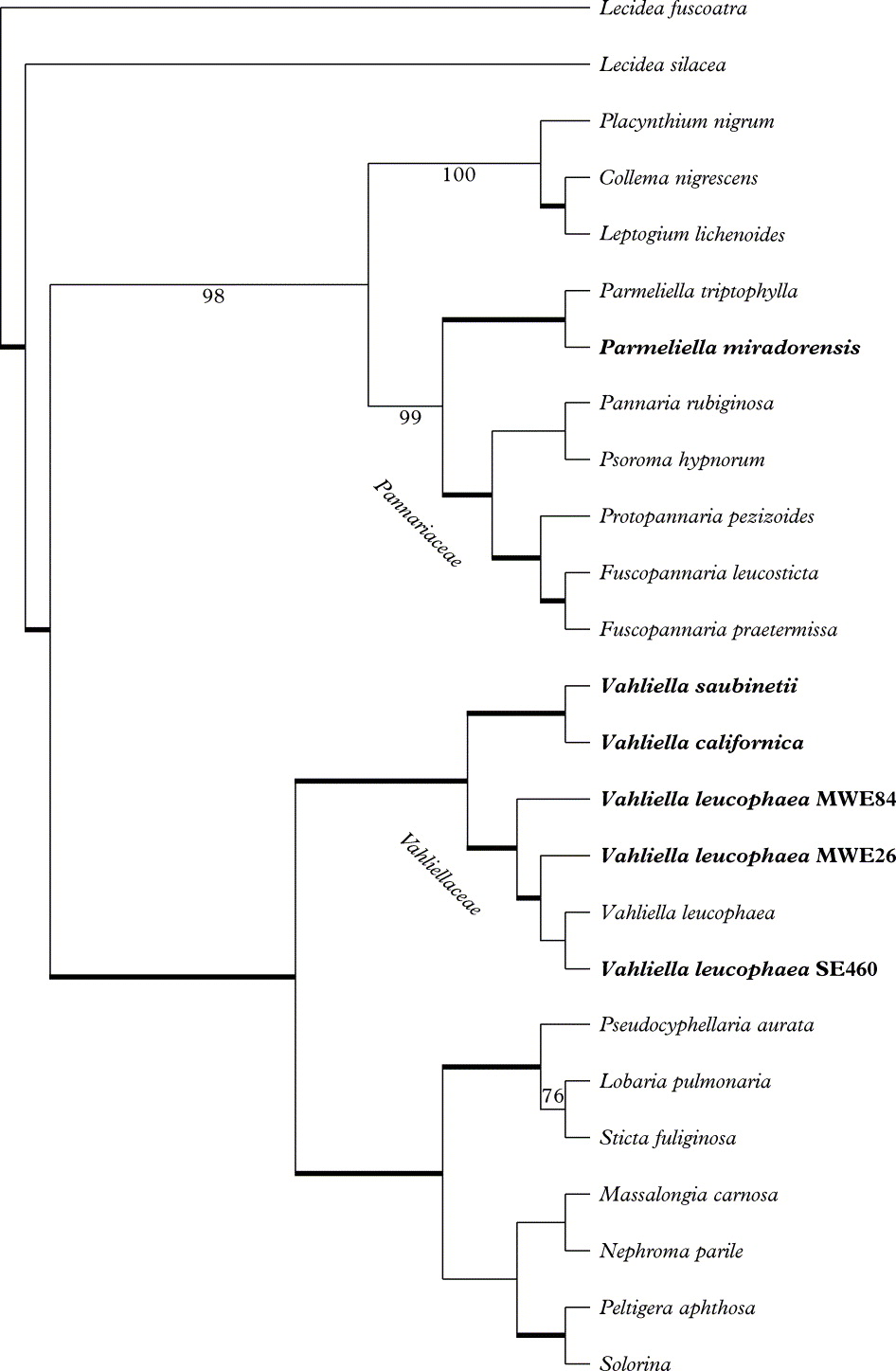
Fig. 1. Phylogenetic relationships among the Peltigerales resulting from Bayesian MCMC analysis (majority-rule consensus tree based on 30 000 trees, ln likelihood = −10648.661±0.169(SE)) based on combined mtSSU rDNA and RPB1 data sets. Samples newly sequenced here are indicated in bold type. Thick internodes received significant support in both types of analysis conducted (Bayesian inference PP ≥95%, MP-BP ≥70%). Nodes supported in the Bayesian inference only are indicated with the PP value below the internode, and one node supported in MP-BS only is indicated with the BS value above the internode. Branch lengths are not indicated, as two distinct branch length sets were allowed.
Discussion
The phylogenies and the well-supported groups recovered correspond very well with topologies retrieved in earlier studies. All samples of Vahliella form one distinct and well-supported monophyletic group (Fig. 1), which is the sister group to one consisting of Lobariaceae (Lobaria, Sticta and Pseudocyphellaria), Massalongiaceae, Nephromataceae, and Peltigeraceae (Peltigera and Solorina). This topology is unlikely to be an artefact caused by poor model assumptions, since our model included mixtures on substitution rate matrices as well as branch lengths. Our study thus confirms that Vahliella is not closely related to the morphologically similar Pannariaceae, where it is currently classified.
Vahliellaceae Wedin, P. M. Jørg. & S. Ekman, fam. nov
Differt a familia Pannariacearum in absentia acidorum lichenicorum, excipulis thallinis male evolutis et ascis cum stratis amyloideis apicalibus.
Typus: Vahliella P. M. Jørg.
Vahliellaceae differs from the related Peltigeraceae, Nephromataceae, and Lobariaceae in general morphology and chemistry, and in ascus structure. Peltigeraceae has a very distinctive apical tube, whereas Nephromataceae has no amyloid apical structure. Lobariaceae has a rather indistinct amyloid layer, which is not very similar to the structure in Vahliellaceae. These three families are all large, foliose lichens, normally with a complex thalline chemistry. Vahliellaceae can be somewhat similar to squamulose members of Massalongiaceae, which also have a somewhat similar ascus structure. Species of Vahliellaceae, however, usually have a distinct bluish black hypothallus that the Massalongiaceae lack. For a detailed discussion of the species of Vahliella, generic characteristics and morphological affinities, see Jørgensen (Reference Jørgensen1994, Reference Jørgensen2008).
This study was supported by The Swedish Research Council (VR 621-2006-3760, 621-2009-5372). We are grateful to the Directors and Curators of the herbaria and museums cited in Table 1 for arranging loans of specimens. The staff at the Molecular Systematics Laboratory assisted with laboratory work at the Swedish Museum of Natural History.