


Introduction
Species traditionally included in the genus Stereocaulon (Stereocaulaceae, lichenized Ascomycota) are characterized by a crustose primary thallus and a fruticose secondary thallus. The primary thallus consists of granules or squamules and is tightly attached to the substratum. The secondary thallus arises from the crustose primary thallus by elongation of thalline tissue and forms so-called pseudopodetia that support phyllocladia or phyllocladoid branchlets, apothecia and in most species cephalodia (Lamb Reference Lamb1951).
In addition to the species forming a dimorphic thallus, six crustose species completely lacking a secondary thallus are included in Stereocaulon. These species are S. cumulatum (Sommerf.) Timdal, S. leucophaeopsis (Nyl.) P. James & Purvis, S. nivale (Follmann) Fryday, S. plicatile (Leight.) Fryday & Coppins, S. tornense (H. Magn.) P. James & Purvis and S. urceolatum (P. M. Jørg.) Högnabba. With the exception of S. urceolatum, the crustose species were transferred to Stereocaulon based on morphological, anatomical and chemical characters (Purvis & James Reference Purvis and James1985; Fryday & Coppins Reference Fryday and Coppins1996; Timdal Reference Timdal2002; Fryday & Glew Reference Fryday and Glew2003). The placement of S. cumulatum, S. leucophaeopsis and S. tornense in Stereocaulon has subsequently been confirmed based on molecular studies (Myllys et al. Reference Myllys, Högnabba, Lohtander, Thell, Stenroos and Hyvönen2005; Högnabba Reference Högnabba2006; see also Printzen & Kantvilas Reference Printzen and Kantvilas2004). Furthermore, S. urceolatum was first placed in the monotypic genus Muhria in the family Stereocaulaceae by Jørgensen & Jahns (Reference Jørgensen and Jahns1987). They had also considered inclusion of the species in Stereocaulon as it has so many characters in common with the genus. However, as the hemiangiocarpic apothecial ontogeny of the species is fundamentally different from the gymnocarpic apothecial ontogeny normally found in Stereocaulon, they concluded that a new genus to accommodate the species was needed. Molecular studies have, however, revealed that the species should be included in Stereocaulon (Myllys et al. Reference Myllys, Högnabba, Lohtander, Thell, Stenroos and Hyvönen2005; Högnabba Reference Högnabba2006; see also Ekman & Tønsberg Reference Ekman and Tønsberg2002; Printzen & Kantvilas Reference Printzen and Kantvilas2004). Hemiangiocarpic apothecial development has also been reported to occur in S. cumulatum and S. tomentosum (Timdal Reference Timdal2002), and occasionally in S. dactylophyllum (Jahns Reference Jahns1970).
Stereocaulon plicatile is a crustose and sorediate species with Porpidia-type asci and submuriform to muriform ascospores. Fryday & Coppins (Reference Fryday and Coppins1996) mention that, apart from the ascospores, S. plicatile is almost impossible to distinguish from S. tornense, but based on the material examined they concluded that S. plicatile tends to have a thinner and less well-developed thallus with more dispersed and flatter areoles. A detailed description of the species is provided by Fryday & Coppins (Reference Fryday and Coppins1996). The species has so far been reported only from the British Isles, where it occurs in montane areas mostly above 800 metres, and from Maine, North America (Fryday Reference Fryday2006). Fryday & Coppins (Reference Fryday and Coppins1996), however, note that it is almost certainly present in other mountainous areas and that it should be looked for particularly in Scandinavia. The inclusion of S. plicatile in the genus is interesting as it is the only species in the genus with submuriform to muriform ascospores. Normally, the species in Stereocaulon have transversely septate ascospores (Lamb Reference Lamb1977; Timdal Reference Timdal2002), although Lamb (Reference Lamb1977) observed that spores may rarely have one to two longitudinal septa.
The phylogenetic position of the crustose species within Stereocaulon is still unresolved. In Myllys et al. (Reference Myllys, Högnabba, Lohtander, Thell, Stenroos and Hyvönen2005), the crustose S. tornense appears as the basal taxon of Stereocaulon, forming the sister taxon to the rest of the genus. The clade including all Stereocaulon species sampled except S. tornense was further divided into two major monophyletic groups, one of which included the remaining crustose species and the other all the fruticose species included in that study. In Högnabba (Reference Högnabba2006), on the other hand, the crustose species formed a monophyletic group that was well nested within Stereocaulon, with the exception of one of the two included specimens of S. cumulatum that appeared as the sister taxon to a clade consisting of all the Stereocaulon specimens included, except the basal S. sorediiferum. The results in Myllys et al. (Reference Myllys, Högnabba, Lohtander, Thell, Stenroos and Hyvönen2005) indicate that the crustose growth form is a plesiomorphy, while the results in Högnabba (Reference Högnabba2006) suggest that the crustose growth form is an apomorphic feature within Stereocaulon.
In order to resolve this ambiguity and to reconsider the phylogenetic position of the species with crustose growth form, we here include two additional crustose species, S. nivale and S. plicatile, in DNA sequence analyses. In the light of the phylogenetic hypotheses obtained, we discuss the evolution of the crustose growth form and the submuriform/muriform spore type. In addition, S. plicatile is reported for the first time from Scandinavia, and its morphological variation is discussed.
Material and Methods
Morphology and chemistry
The morphology of the S. plicatile material from Sweden was studied using standard light microscope techniques. Spore measurements were made on material mounted in water. Extrolites were detected using thin-layer chromatography (TLC) as described in Orange et al. (Reference Orange, James and White2001).
DNA extractions, amplification and sequencing
Total DNA was extracted using DNeasy® Plant Mini Kit (Qiagen) or DNeasy® Blood and Tissue Kit (Qiagen) according to the protocols enclosed with the kits, except that the liquid nitrogen phase was omitted for both kits. Instead, the thallus fragments were ground with a mini-pestle in a small amount of lysis buffer provided with the kits. The DNA extracted was eluted in 160 µl of elution buffer when the Plant Mini Kit was used, and 100 µl of elution buffer when the Blood and Tissue Kit was used. The extraction products were used undiluted in the PCR reactions.
To amplify the nITS rDNA region, the primer pairs ITS1F (Gardes & Bruns Reference Gardes and Bruns1993) and ITS4 (White et al. Reference White, Bruns, Lee, Taylor, Innis, Gelfand, Sninsky and White1990) or ITS1-LM (Myllys et al. Reference Myllys, Lohtander, Källersjö and Tehler1999) and ITS2-KL (Lohtander et al. Reference Lohtander, Myllys, Sundin, Källersjö and Tehler1998) were used. The partial beta-tubulin gene was amplified using the primer pair Bt3-LM and Bt10-LM (Myllys et al. Reference Myllys, Lohtander and Tehler2001). PCR amplification was undertaken using illustra PuReTaq Ready-To-Go™ PCR Beads (GE Healthcare). The 25 µl PCR samples were prepared by adding 1 µl of each primer at 10 µM concentration, 4 or 5 µl of template DNA and 18–19 µl sterile water to the 0·2 ml tubes with PCR beads. PTC-100, PTC-200 Thermal Cyclers (MJ Research) and Mastercycler® ep gradient S (Eppendorf) were used to perform the PCR cycles. Slightly different settings were successfully used in the PCR reactions. ITS1F+ITS4: 95°C for 5 min; 5 cycles of 30 s at 95°C, 30 s at 55°C, 60 s at 72°C; 30 cycles of 30 s at 95°C, 30 s at 52 or 53°C, 60 s at 72°C; 7 min at 72°C (omitted for some of the reactions). ITS1F+ITS4 and ITS1LM+ITS2KL: 95°C for 5 min; 5 cycles of 30 s at 95°C, 30 s at 58°C, 60 s at 72°C; 30 cycles of 30 s at 95°C, 30 s at 56°C, 60 s at 72°C; 7 min at 72°C; Bt3-LM+Bt10-LM: 95°C for 5 min; 5 cycles of 30 s at 95°C, 30 s at 55 or 56°C, 60 s at 72°C; 30 cycles of 30 s at 95°C, 30 s at 52 or 54°C, 60 s at 72°C; 7 min at 72°C. The PCR products were purified using illustra GFX PCR DNA and Gel Band Purification Kit (GE Healthcare) following the manufacturer's instructions. Purified DNA was eluted with 30–50 µl sterile water (elution buffer 6) included in the kit. The amount of sterile water used depended on the strength of the product when visually observed on an agarose gel.
For sequencing of the PCR products, BigDye® Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems) was used. Sequencing samples containing 3 µl BigDye, 1 µl primer at 2·5 µM concentration, 2 µl purified PCR product and 4 µl sterile water were prepared. The primers used for sequencing were the same as listed above for the PCR amplification. The samples were run using the following settings: 96°C for 30 s, 50°C for 15 s, and 60°C for 4 min. The same equipment as in the PCR reactions was used to run the sequencing reactions. For the post-reaction purification of the samples, the protocol described in Högnabba (Reference Högnabba2006) was followed. Sequencing of these samples was made by an ABI prism 377 automatic sequencer. Sequencing of the remaining samples was carried out at Macrogen Inc. (http://www.macrogen.com). The DNA concentration of the PCR products sequenced at Macrogen was measured using a BioPhotometer (Eppendorf) and UVette® cuvettes (Eppendorf) using the 10 mm optical path length. 50 µl dilutions of the PCR products were prepared for the concentration measurements by adding 7 µl of the PCR products and 43 µl sterile water.
SeqMan II version 4.0 (DNASTAR) was used to assemble the sequences obtained. IUPAC ambiguity codes were used when base calling was equivocal. BLAST searches were used to confirm the sequence identity. All the new sequences were homologous (>94% of identity) with Stereocaulon sequences in GenBank.
Taxon sampling
To study the position of the crustose species of Stereocaulon, a dataset based on that published in Högnabba (Reference Högnabba2006) was constructed. Representatives from all the major groups recognized in that study were selected (Table 1). Of the fruticose species, only specimens where both ITS and beta-tubulin sequences were available were selected, with the exception of S. sorediiferum which was the basal taxon of the genus in Högnabba (Reference Högnabba2006), forming the sister group to the rest of the genus. For this taxon, only the beta-tubulin sequence was available despite several attempts to sequence the ITS regions. The ITS sequence of S. delisei was newly sequenced for this study. For the crustose members of the genus, all sequences available were included in the analyses. Sequences of S. nivale (two specimens) and S. plicatile were produced for the first time for this study. In addition, the beta-tubulin sequence of one new S. leucophaeopsis specimen was included, and the ITS region was successfully sequenced for one of the S. urceolatum specimens for which only the beta-tubulin sequence was included in Högnabba (Reference Högnabba2006). Altogether, eight new sequences of the crustose Stereocaulon specimens were included in this study. Species from the genus Lepraria were selected as outgroup taxa. The genus Lepraria is included in the family Stereocaulaceae (Lumbsch & Huhndorf Reference Lumbsch and Huhndorf2010) and has been shown to form the sister group of Stereocaulon (Myllys et al. Reference Myllys, Högnabba, Lohtander, Thell, Stenroos and Hyvönen2005; Miądlikowska et al. Reference Miądlikowska, Kauff, Hofstetter, Fraker, Grube, Hafellner, Reeb, Hodkinson, Kukwa and Lücking2006). We also made preliminary analyses including more distantly related taxa from the Cladoniaceae and the Lecanoraceae. In all analyses, Lepraria and Stereocaulon formed a sister group. In the final analyses we therefore chose to include only Lepraria as outgroup due to alignment problems of the ITS regions when more distantly related taxa were included. The final dataset consisted of 37 specimens of 31 species from the ingroup, including all crustose species currently classified in Stereocaulon. Three specimens representing two taxa of the outgroup were included in the final analyses.
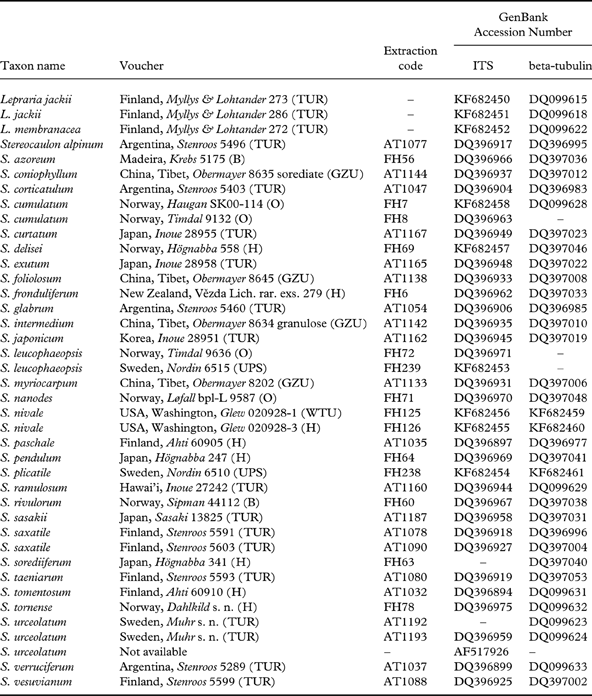
Table 1. Specimen information and GenBank accession numbers for sequences of Stereocaulon and outgroup species analyzed in this study. New sequences are indicated by accession numbers in bold.
Sequence alignment and phylogenetic analyses
We used four different approaches for analyzing the data: conventional parsimony, parsimony with direct optimization, maximun likelihood (ML) and Bayesian inference. In the conventional parsimony approach, ML and the Bayesian methods, the sequences were aligned prior to the analyses. The ITS regions showed considerable length variation and the included beta-tubulin intron region showed minor length variation. Alignments (available by contacting the corresponding author) of these regions were made with the software MUSCLE version 3.7 (Edgar Reference Edgar2004). Ambiguously aligned sites of the ITS regions were removed manually. The 5.8S region that was aligned together with the ITS regions showed no length variation. The exons of the beta-tubulin sequences were all of equal length and could be aligned manually without problems. In order to examine putative conflicts among the loci, bootstrap analysis of the alignment of ITS regions and beta-tubulin separately was carried out. Each clade with bootstrap support >75% was scanned, according to the method described by Lutzoni et al. (Reference Lutzoni, Kauff, Cox, McLaughlin, Celio, Dentinger, Padamsee, Hibbett, James and Baloch2004). No conflicts were found and the alignments were concatenated. The conventional parsimony analysis was made with the program TNT (Goloboff et al. Reference Goloboff, Farris and Nixon2008). A traditional search with the following settings was performed: starting trees obtained with 1000 replicates, TBR branch-swapping algorithm, saving 20 trees per replicate, gaps were treated as a 5th character state. Jackknife support values (Farris et al. Reference Farris, Albert, Källersjö, Lipscomb and Kluge1996) for the nodes were calculated with 1000 replicates using the program TNT.
Length variation in the non-coding regions such as the ITS often causes problems with homology assumptions, and alignments of such regions often include ambiguously aligned sites. To overcome the alignment problems caused by the length variation in the ITS regions and the beta-tubulin intron region, we performed direct optimization (Wheeler Reference Wheeler1996) as an alternative to the conventional parsimony approach. In the direct optimization procedure the homology assumptions and the tree search are made simultaneously, without the need to align the sequence prior to analysis. In the analyses, direct optimization was performed for the ITS and the beta-tubulin intron regions, while the 5.8S region and the beta-tubulin exon regions were treated as prealigned (i.e., left as fixed alignments within this analysis). The analysis was performed using version 1.4.2.1 of the program POY (Varón et al. Reference Varón, Vinh, Bomash and Wheeler2008, Reference Varón, Vinh and Wheeler2010) running on the Vuori HP CP4000 BL ProLiant supercluster at the IT Center for Science, Espoo, Finland. Initially, 1000 random-addition trees were generated with transitions, transversions, and indels given equal weight. The initial trees were refined using SPR and TBR branch swapping. In addition to the optimal trees, suboptimal trees up to 10% longer than the optimal trees were evaluated during the SPR and TBR swapping. This was followed by 15 rounds of ratcheting (Nixon Reference Nixon1999) in which 20% of the characters were re-weighted randomly by a factor of three. Finally, 200 iterations of tree-fusing (Goloboff Reference Goloboff1999) were performed. All optimal and unique trees were retained between the different searches. The commands used in the POY analysis are provided in Appendix 1. For each of the most parsimonious trees found, separate implied alignments of the regions subjected to direct optimization were obtained. The implied alignments were concatenated with the alignments of the regions without length variation using the program MacClade (version 4.06; Maddison & Maddison Reference Maddison and Maddison2003). The matrices obtained were used for calculating jackknife support values (Farris et al. Reference Farris, Albert, Källersjö, Lipscomb and Kluge1996) for the clades. Separate jackknife analyses were run for each matrix. If the analyses resulted in different support for a clade, the lowest and highest values are indicated. The jackknife support values were calculated in 1000 replicates using the program TNT (Goloboff et al. Reference Goloboff, Farris and Nixon2008).
The strict consensus of the most parsimonious trees retained in the conventional parsimony analysis was calculated using the software Mesquite version 2.74 (Maddison & Maddison Reference Maddison and Maddison2010). In the direct optimization analysis, the strict consensus of the retained trees was exported at the end of the POY analysis.
We used maximum likelihood analysis as implemented in RAxML 7.04 (Stamatakis Reference Stamatakis2006), using GTRGAMMA model with four partitions (ITS and each codon position of beta-tubulin). All the parameters were estimated from the data. The clade support was estimated with 500 replicates of ‘fast bootstrap’. Bayesian analysis was carried out in MrBayes 3.1.2 (Huelsenbeck & Ronquist Reference Huelsenbeck and Roquist2001) with default priors. The best-fit evolutionary model to each loci was selected by MrModeltest (Nylander Reference Nylander2004) under AIC criterion. The GTR+G and GTR+I+G models were selected for beta-tubulin and ITS regions, respectively. The posterior probabilities were approximated by sampling trees using Markov Chain Monte Carlo (MCMC). Two simultaneous runs with 10 000 000 generations, each starting with a random tree and employing 4 simultaneous chains, were executed. Every 1000th tree was saved into a file. The convergence of the chains was determined with TRACER 1.5 (Rambaut & Drummond Reference Rambaut and Drummond2007) and a standard deviation of split frequencies was <0·005 between the runs. The potential scale reduction factor (PSRF) was 1·0 for all the parameters. The first 1 000 000 generations were deleted as the ‘burn in’ of the chain and the 50% majority-rule consensus tree was calculated using the ‘sumt’ command in MrBayes.
The trees were visualized using the software FigTree version 1.3.1 (Rambaut Reference Rambaut2009), and the graphical appearance of the trees shown was further edited using the program Inkscape version 0.47preI r21720 (http://www.inkscape.org).
Results
Stereocaulon plicatile (Leight.) Fryday & Coppins in Scandinavia
A crustose Stereocaulon species with submuriform to muriform spores was found by A. Nordin during fieldwork in Jämtland, Sweden in 2007. The species found in Jämtland contained atranorin and stictic acid and thereby has the same chemistry as reported for S. plicatile. Some of the other characteristics of the Swedish material, however, differ slightly to what was reported for S. plicatile by Fryday & Coppins (Reference Fryday and Coppins1996). The size of the ascospores of the Swedish material was found to be 22·6–[30·0]–35·0×9·0–[11·7]–14·7 µm (mean values in brackets, n=17) compared to 20–32×10–15 µm reported for S. plicatile. The mean values of the length and width of the spores in the Swedish material, however, fall within the ranges reported for the species. The apothecia in the Swedish material are up to 3 mm wide while the apothecia in S. plicatile were reported to be up to 1·6 mm wide. Finally, the hymenium in the Swedish material seems to be thicker than 110 µm, as was mentioned in the species description of S. plicatile. The Swedish material was found in an old copper mine, whereas S. plicatile was reported to occur on siliceous rocks and pebbles, and no possible copper association was mentioned. Cephalodia were reported to be absent in S. plicatile. However, the material collected in Sweden is associated with Stigonema. This kind of loose association with cyanobacteria (attached to the thallus, but not confined to cephalodia) is also reported for the other crustose Stereocaulon species (Jørgensen & Jahns Reference Jørgensen and Jahns1987; Jahns et al. Reference Jahns, Klöckner, Jørgensen and Ott1995; Timdal Reference Timdal2002). Furthermore, when compared to the material of S. plicatile collected by Fryday (no. 5609, UPS), the general appearance of the Swedish material is very similar. Interestingly, this specimen is also associated with Stigonema. In our opinion, it is unlikely that there are two crustose Stereocaulon species with submuriform to muriform spores and the same chemistry that would be morphologically so similar. Therefore, our conclusion is that the Swedish material belongs to S. plicatile, but the variation of some of the morphological characteristics is slightly wider than reported by Fryday & Coppins (Reference Fryday and Coppins1996), who actually stated that the species should particularly be looked for in Scandinavia. However, until molecular data from British material has been obtained for comparison, the relationship between the populations remains uncertain. The Scandinavian population probably extends to adjacent areas in Norway, and might even be distributed along the Scandinavian mountains further north and reach northern Finland.
Specimens examined. Sweden: Jämtland: Åre parish, Handöl copper mine just W of the road between Handöl and Storulvån, c. 1 km S of Handöl, 63°14′45·8″N, 12°25′47·3″E (WGS84), alt. 640 m, on rock in an open shaft close to the road, 2007, A. Nordin 6510 (UPS); on loose pieces of rock in the N-most open shaft, 2008, A. Nordin 6677 (UPS); on rockwall in the E-most open shaft, 2008, A. Nordin 6680 (UPS).—Great Britain: Scotland: V.C. 97, West Inverness-shire: Creag Meagaidh, Grid 27/405,871, alt. 1000 m, on siliceous pebbles above area of prolonged snow-lie, NW of summit, 1994, A. M. Fryday 5609 (UPS).
Phylogenetic analyses
The concatenated alignment contained 1210 positions (454 of ITS regions and 756 of beta-tubulin), of which 824 were constant (285 of ITS regions and 539 of beta-tubulin) and 276 were parsimony-informative (116 of ITS regions and 160 of beta-tubulin). The conventional phylogenetic analysis resulted in 78 equally parsimonious trees with a length of 952 steps. The direct optimization analysis resulted in four equally parsimonious solutions with a length of 1459 steps. The tree lengths from the analyses are not comparable as the ambiguously aligned sites were removed from the alignment before it was used in the conventional analysis, and therefore fewer characters were included. The strict consensus tree of the direct optimization analysis is somewhat more resolved than the strict consensus tree of the conventional phylogenetic analysis. However, in both phylogenies five major, highly supported clades can be recognized. These are: 1) S. cumulatum (jackknife=100/100), 2) S. urceolatum (j=99/99–100), 3) the crustose Stereocaulon species except S. cumulatum and S. urceolatum (j=98/99–100), 4) subgenus Holostelidium (j=85/89–91), and 5) subgenus Stereocaulon (j=98/99–100). The clades and support values are indicated in the strict consensus of the trees found in the direct optimization analysis (Fig. 1). The results from the parsimony analyses are essentially the same and therefore we only show the direct optimization tree. The ML analyses yielded a tree with the likelihood value of LnL=−6059·93, while the likelihood for the Bayesian tree was LnL=−6327·26. The ML and Bayesian topology showed five well-supported clades similar to those in the parsimony analyses. Figure 2 shows the 50% majority-rule consensus tree of the Bayesian analysis.
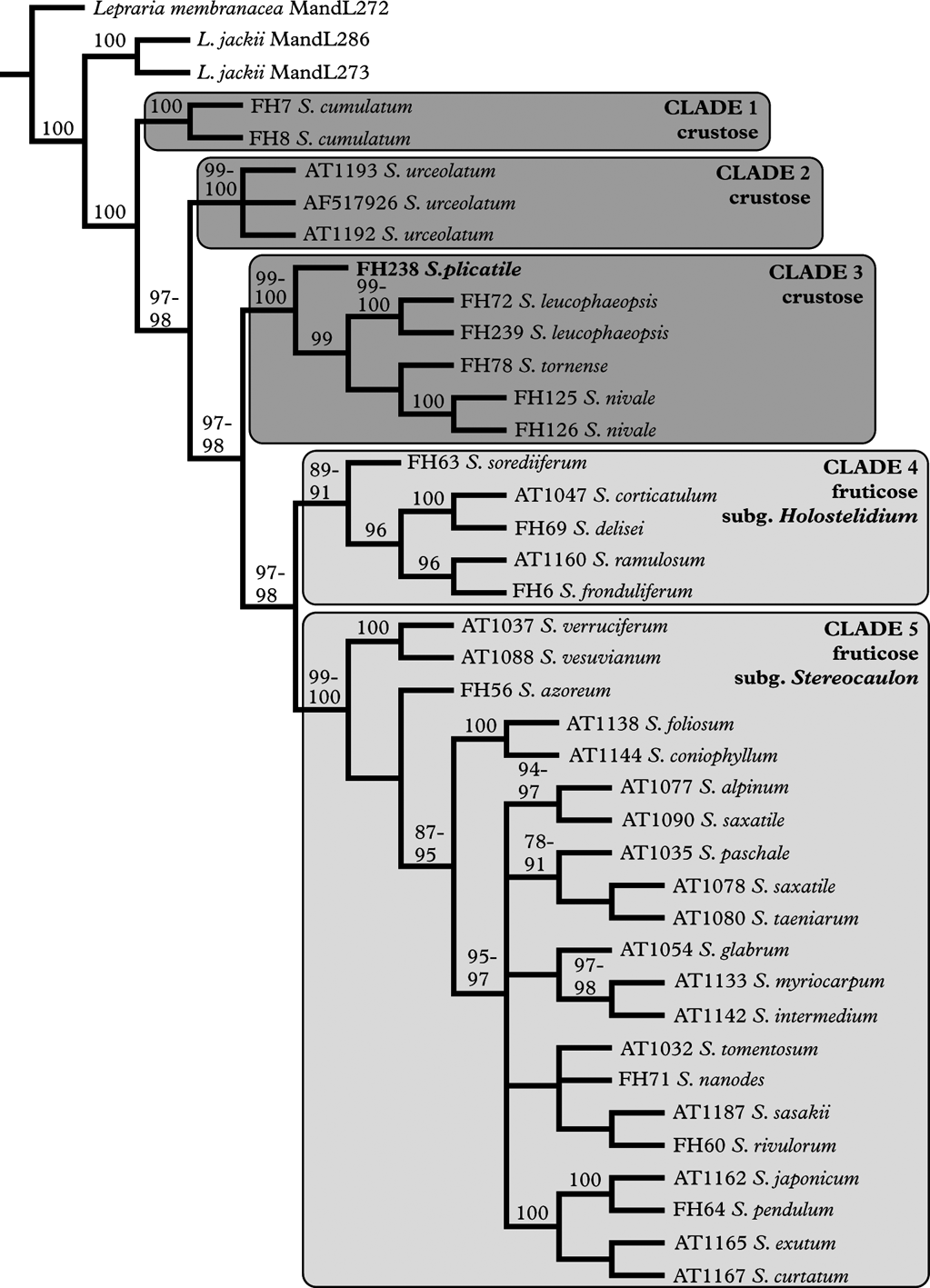
Fig. 1. Phylogenetic relationships among Stereocaulon species with emphasis on the crustose taxa. The strict consensus of four equally parsimonious trees for Stereocaulon species found in the direct optimization procedure. Jackknife support values are indicated above the nodes when ≥70. For the nodes where the support value differed between the four jackknife analyses, based on the different implied alignments, the highest and lowest supports obtained are indicated. Main clades, growth form, current classification for the fruticose species and extraction codes are indicated. The placement of S. plicatile with submuriform to muriform spores is indicated in bold.
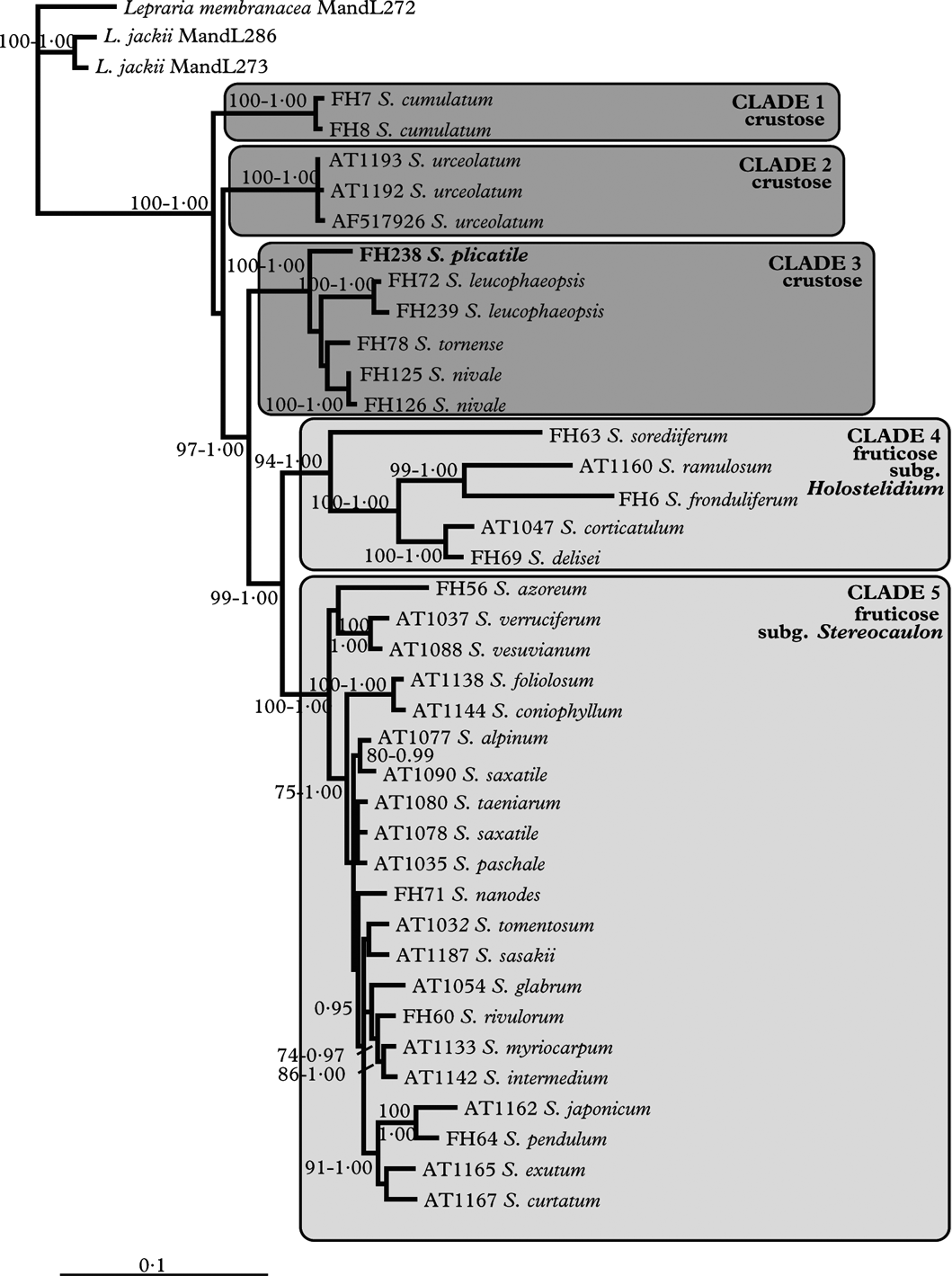
Fig. 2. Phylogenetic relationships among Stereocaulon species with emphasis on the crustose taxa. The 50% majority-rule consensus tree of a Bayesian analysis of nrITS and beta-tubulin of Stereocaulon species. Branch support is shown where bootstrap ≥70% for ML and posterior probability ≥0·95.
The relationships between the major groups are identical in the results obtained from the four analyses, with the exception that S. cumulatum is the basal taxon of the genus in the strict consensus obtained from direct optimization, ML and Bayesian analyses, while in the strict consensus from the conventional parsimony analysis a trichotomy is formed at the base of Stereocaulon, leaving the basal taxon of the genus unresolved. In the clades comprising more than one species, the relationships between the species are identical in clade 4 in all analyses, while the phylogenetic relationships within clades 3 and 5 differ to some extent between the trees. In clade 3, S. plicatile forms the basal taxon of the clade in all analyses. In the consensus tree of the conventional parsimony analysis, the relationships between S. leucophaeopsis, S. nivale and S. tornense are not resolved, while in the other analyses S. leucophaeopsis is the sister taxon to a group formed by S. nivale and S. tornense. Clade 5 is highly unresolved in the different analyses and the positions of some taxa differ between the analyses.
Discussion
Our results imply a basal position of the crustose Stereocaulon species in the genus and that the crustose species do not form a monophyletic entity. The basal position of the crustose species suggests that the crustose growth form is the plesiomorphic feature of the genus. Our results show that the crustose S. plicatile and S. nivale, which are incorporated in molecular analyses for the first time, are included in a strongly supported clade together with the crustose S. leucophaeopsis and S. tornense. Thus, the inclusion of these two taxa in the genus is also supported by DNA sequence data. The division of the subgenera Holostelidium and Stereocaulon into two clades supports the recognition of the two subgenera.
The basal position of the crustose species is supported by the conventional parsimony, the direct optimization, ML and Bayesian analyses. However, in the trees from the direct optimization, ML and Bayesian analyses, S. cumulatum is the basal taxon forming the sister species to the rest of the genus and S. urceolatum forms the sister taxon to the monophyletic group formed by the clades 3–5. In the conventional parsimony analysis, a trichotomy is formed at the base of the genus, and the relationship between S. cumulatum and S. urceolatum.
The basal position of crustose Stereocaulon species is in accordance with the results presented in Myllys et al. (Reference Myllys, Högnabba, Lohtander, Thell, Stenroos and Hyvönen2005). However, in that study, S. tornense formed the basal taxon of the genus. In the results presented here, S. tornense is nested in clade 3 together with the crustose S. leucophaeopsis, S. nivale and S. plicatile. In the combined analysis in Myllys et al. (Reference Myllys, Högnabba, Lohtander, Thell, Stenroos and Hyvönen2005), S. cumulatum and S. urceolatum (as Muhria urceolata) formed a clade with S. leucophaeopsis. This grouping is not observed in the present study. In Högnabba (Reference Högnabba2006), all the crustose Stereocaulon species, with the exception of one specimen of S. cumulatum (FH8), formed a clade that was nested within the genus. This relationship suggested that the crustose species formed a lineage in which the fruticose growth form has been lost, or is currently not expressed. Plausible explanations for the observed differences in the results of the previous and present studies are character and/or taxon sampling. In the present study, we included one intron region of the beta-tubulin sequences that was excluded in Myllys et al. (Reference Myllys, Högnabba, Lohtander, Thell, Stenroos and Hyvönen2005). The intron may have brought additional information to the analyses. Furthermore, in Myllys et al. (Reference Myllys, Högnabba, Lohtander, Thell, Stenroos and Hyvönen2005), the focus was on the family level and therefore the selection of loci was different (ITS not used). The loci used in the present study and in Högnabba (Reference Högnabba2006) are identical. The differences observed in the position of the crustose species are therefore most probably a result of the more extensive taxon sampling of the crustose species presented here. The absence of monophyly of S. cumulatum in Högnabba (Reference Högnabba2006) is probably due to stochastic errors in the analyses. The specimen of S. cumulatum (FH7) was represented only by the beta-tubulin sequence, and the other specimen of S. cumulatum (FH8) was represented only by the ITS sequence. In the present work, both specimens have ITS sequences and they are identical. Therefore, the current relationship is more robust than in the previous phylogenies. The crustose morphology was suggested to be the ancestral growth form in Stereocaulon by Lamb (Reference Lamb1951). Assuming that Lamb's hypothesis was correct, Fryday & Glew (Reference Fryday and Glew2003) suggested that S. nivale would occupy a basal position in Stereocaulon. Our results support the hypothesis postulated by Lamb (Reference Lamb1951), but not the basal position of S. nivale as suggested by Fryday & Glew (Reference Fryday and Glew2003).
The position of S. plicatile and S. nivale in clade 3 is supported by all analyses. The inclusion of these two crustose taxa in Stereocaulon that was based on morphological and chemical characters is thus clearly supported also by DNA sequence data, and their inclusion in Stereocaulon is thereby well founded. The inclusion of S. plicatile, with submuriform to muriform spores, is particularly interesting. The nested position of S. plicatile in Stereocaulon suggests that the submuriform to muriform spore type is an autapomorphic character that has been gained independently within the genus.
Timdal (Reference Timdal2002) tabulated distinguishing features of the crustose Stereocaulon species, excluding S. nivale which was characterized and transferred to Stereocaulon later (Fryday & Glew Reference Fryday and Glew2003). Some of the features listed in these works separate clade 3 from the more basal S. cumulatum and S. urceolatum: the maximum areole diameter is 0·5–1·0 mm (vs. 4 and 5 mm for S. cumulatum and S. urceolatum, respectively), the apothecia are gymnocarpic (vs. hemiangiocarpic in the basal species), and the maximum apothecium diameter is 1·4–1·6 mm (vs. 0·5 mm in the basal species). In Scandinavian S. plicatile, the apothecia are even wider (see species description). Thus, the well-supported clade 3 can also be recognized based on morphological characters, which opens up the possibility of treating the group as a taxonomic entity, preferably as a subgenus. Furthermore, S. cumulatum and S. urceolatum that form clades 1 and 2, respectively, should then be included in separate monotypic subgenera. However, this taxonomic treatment is not done here. The few studies including crustose Stereocaulon species are in contradiction (see above) and further studies including additional data, more loci in particular, are necessary to reach phylogenetic stability, based on which well-founded taxonomic decisions can be made. Also, inclusion of additional fruticose taxa, especially of the subgenus Holostelidium, which are under-represented in the analyses published so far, is necessary.
The fruticose species form two major clades. Clade 4 corresponds to the subgenus Holostelidium and clade 5 to the subgenus Stereocaulon. This result is essentially in accordance with the results in Högnabba (Reference Högnabba2006), where the subgenus Holostelidium formed a monophyletic group with the exception of S. sorediiferum, which was the basal species of the genus. Also in that analysis, the subgenus Stereocaulon was monophyletic with the exception of S. pileatum, S. virgatum and one specimen of S. verruciferum. This can be explained by the fact that, for these three specimens only, the ITS regions were included in the analyses presented in Högnabba (Reference Högnabba2006). One specimen of S. verruciferum, for which the beta-tubulin sequence was also included, was clearly nested in the subgenus Stereocaulon.
The subgenus Holostelidium is characterized by the so-called holostelidious pseudopodetia development, in which the pseudopodetia are formed by elongation of all layers of the primary thallus. In the subgenus Stereocaulon, the pseudopodetia are formed by elongation of the basal medullary layer of the primary thallus (i.e. enteropodious pseudopodetia development). As the two subgenera form distinct groups, the pseudopodetia development seems to be a good feature with which to separate them. This is perhaps not a surprise as the two types of pseudopodetia development are essentially different (Lamb Reference Lamb1951).
Within the subgenus Holostelidium, the relationships between the species are identical in all analyses. Also, the relationships within subgenus Holostelidium found in the present study are in accordance with the results in Högnabba (Reference Högnabba2006), with the exception of S. sorediiferum as mentioned above. The contradictory placement of S. sorediiferum needs further investigation. Interestingly, S. sorediiferum is included in the section Holostelidium, while all other species of the subgenus Holostelidium included here, as well as in Högnabba (Reference Högnabba2006), are placed in the section Aciculisporae by Lamb (Reference Lamb1977). Within the subgenus Stereocaulon, the relationships between the taxa differ between the conventional and the direct optimization methods. This may be because ambiguously aligned sites were discarded prior to the conventional analyses. The clades found within the subgenus Stereocaulon in the direct optimization tree correspond in most cases to clades found within the corresponding group in Högnabba (Reference Högnabba2006). However, to reach final conclusions about phylogenetic relationships of the fruticose species, further studies including broader sampling of taxa and loci are required.
We thank P. Tahvanainen and M. Piisilä for assistance in the laboratory, J. Hyvönen for valuable advice on the phylogenetic analyses and T. Ahti for comments on the manuscript. LM and SS were supported by the Academy of Finland (grants 133858, 211172, 124203) and FH by the Oskar Öflund Foundation. The Willi Hennig Society is acknowledged for making the program TNT freely available.
Appendix 1. Search options used in the POY analysis. For explanations of the commands see Varón et al. (Reference Varón, Vinh, Bomash and Wheeler2008)
The following commands were used in the direct optimization analysis:
read("CrustSter28its1.fas","CrustSter28its3.fas","CrustSter28BTint.fas")
read(prealigned:("CrustSter28its2.fas",tcm:"111.txt"))
read(prealigned:("CrustSter28bt1.fas",tcm:"111.txt"))
read(prealigned:("CrustSter28bt2.fas",tcm:"111.txt"))
set(seed:1,log:"CrustSter28.log",root:"LeprolomamembranaceumMandL272")
transform((all,tcm:"111.txt"))
build(1000)
swap(threshold:10)
select()
perturb(transform(static_approx),iterations:15,ratchet:(0.2,3))
select()
fuse(iterations:200,swap())
select()
report(asciitrees)
report("CrustSter28graphtrees",graphtrees)
report("CrustSter28graphstrictconsensus",graphconsensus)
report("CrustSter28trees.txt",trees)
report("CrustSter28strictconsensus.txt",consensus)
report("CrustSter28stats.txt",treestats)
report("CrustSter28impliedalignment",ia:names:("CrustSter28its1.fas","CrustSter28its3.fas","CrustSter28BTint.fas"))
report("CrustSter28.time",timer:"total time")
set(nolog)
exit()



