


In the course of our studies of lichenicolous fungi, we have been seeking a quick and reliable method for extracting DNA from minute specimens and microscopic material. Extraction of fungal genomic DNA generally involves breaking the hyphal walls and then extracting and purifying the genomic DNA. Commercial extraction kits (e.g. QIAGEN DNeasy Plant Mini Kit) or the classic CTAB protocol (Grube et al. Reference Grube, DePriest, Gargas and Hafellner1995; Cubero et al. Reference Cubero, Crespo, Fatehi and Bridge1999) are generally used. Although these techniques provide DNA of a satisfactory quantity and quality, most are tedious and time-consuming and involve the use of hazardous chemicals in the extraction process (e.g. phenol, chloroform, isoamyl alcohol). Moreover, commercial kits are relatively expensive and inappropriate for minute samples which can be easily lost during processing.
For our research, we required a method for extracting genomic DNA from lichenicolous and lichen-forming fungi that would be less time consuming, cheap, not hazardous, and suited for use with the large number of samples necessary to thoroughly investigate phylogenetic relationships in these fungi. Here we report a relatively simple thermolysis method for extracting fungal genomic DNA, and the testing of its efficacy by amplification, in this case of the nrDNA LSU and ITS regions.
Direct PCR as proposed by Wolinski et al. (Reference Wolinski, Grube and Blanz1999) is commonly used for minute samples (e.g. Lawrey et al. Reference Lawrey, Binder, Diederich, Molina, Sikaroodi and Ertz2007; Ertz et al. Reference Ertz, Lawrey, Common and Diederich2014) to avoid an extraction step. It is not, however, always easy to use for lichenicolous fungi intimately associated with the host, or appropriate if a stock of extracted DNA is required for future studies. Moreover, direct PCR could be unsuitable for strongly melanized material or in the presence of PCR inhibitors (Eckhart et al. Reference Eckhart, Bach, Ban and Tschachler2000; Schrader et al. Reference Schrader, Schielke, Ellerbroek and Johne2012).
There is, however, a promising and rapid technique based on the thermolysis of the sample in the presence of a chelating resin (Walsh et al. Reference Walsh, Metzger and Higuchi1991) that could be suitable. It has been successfully applied in a wide range of eukaryotic organisms (e.g. Pedersen et al. Reference Pedersen, Russell, Newton and Ansell2006; Strange et al. Reference Strange, Knoblett and Griswold2009; HwangBo et al. Reference HwangBo, Son, Lee, Min, Ko, Liu and Jeong2010; Casquet et al. Reference Casquet, Thebaud and Gillespie2012) including some non-lichenized fungi (e.g. Zhang et al. Reference Zhang, Zhang, Liu, Wen and Wang2010; Turan et al. Reference Turan, Nanni, Brunelli and Collina2015). In order to explore the efficacy of this method, it was necessary to customize the technique so it could be used with limited amounts of material and yet produce DNA suitable for PCR amplification.
Chelex® 100 (Bio-Rad, Hercules, CA, USA) is a specialized resin that chelates metal ions (Chelex=Chelating Ion Exchange Resin), supplied in the form of fine crystals composed of styrene divinylbenzene co-polymers containing paired iminodiacetate ions which act as chelating groups. The alkalinity of Chelex suspensions (pH 10–11) and exposure to 100°C results in the disruption of membranes and denaturation of the DNA. Moreover, it prevents the degradation of DNA by chelating metal ions that otherwise act as catalysts in DNA breakdown (Singer-Sam et al. Reference Singer-Sam, Tanguay and Riggs1989). Furthermore, Chelex is known to remove PCR inhibitors such as mineral ions and humic acids (Tsai & Olson Reference Tsai and Olson1992; Tebbe & Vahjen Reference Tebbe and Vahjen1993). We tested the method using the following procedures.
Extraction. The fresh and dried reference material used in our evaluation is detailed in Table 1. Samples were removed under a dissecting microscope using a razor blade, forceps and an acupuncture needle as follows: (a) a small piece (c. 1–2 mm2) of the young lichen lobule (free of any visible lichenicolous fungi); (b) one ascoma of lichenicolous fungi (free of host tissue); or (c) a few hyphae in the case of lichenicolous fungi lacking ascomata, taking care to exclude host tissue. The samples were placed in a 1·5 ml Eppendorf tube with 15 μl of sterile distilled water. The thallus fragments and ascomata were then macerated with a sterile lancet. The samples were stored at −20°C for 12–48 h before processing.
Table 1 Voucher information for specimens used in this study together with the concentration of extracted DNA and its purity. The values in parentheses correspond to the 1/10 dilution of the extract. The optimal purity of DNA is highlighted in bold
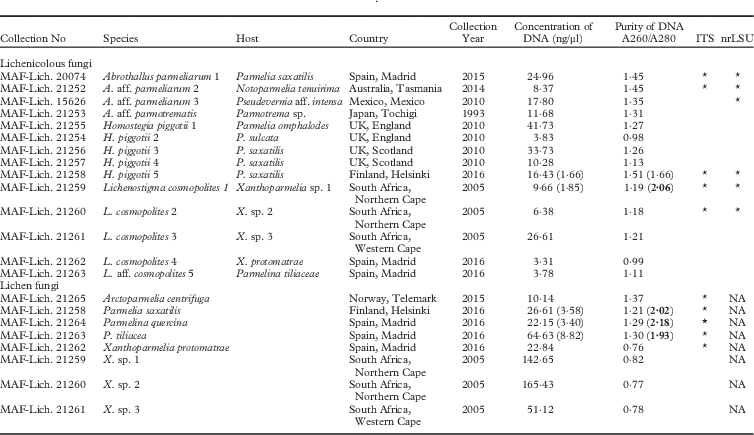
*=positive PCR and clean DNA sequence acquired; NA=not analyzed. The sequences obtained will be presented in separate publications.
A 10% Chelex solution was prepared in a 1·5 ml Eppendorf tube using 100 μg Chelex® 100 Resin (1421253, molecular biology grade, 200–400 mesh, sodium form, 50 g; Bio-Rad, Hercules, CA, USA) and 1 ml of sterile distilled water. Before pipetting, the 10% Chelex solution was mixed to prevent the resin becoming deposited on the bottom of the reaction tube, and to avoid the final concentration varying in different parts of the tube. 100 μl of 10% Chelex solution was added directly to the tube with the sample. The samples were vortex mixed for 10 s and then incubated in a Multiplaces thermostat dry block (J. P. Selecta, Barcelona, Spain) at 95°C for 20 min. They were briefly shaken or vortexed every 5 min during this time to prevent the resin depositing in the bottom of the tube. The tubes were then centrifuged at 14000 rpm for 30s and the supernatant (50–70 μl) was carefully removed, taking care to avoid contact with the resin, and transferred to a clean 1·5 ml tube. The samples were either frozen (at −20°C) or used directly for PCR.
DNA quantification and quality. DNA sample concentrations and purity were determined spectrophotometrically using a NanoDrop ND-1000 Spectrophotometer (Isogen Life Science, De Meern, The Netherlands) at the Unidad de Genómica (Parque Científico de Madrid). Concentration results are given in ng/μl, and the DNA purity results are reported as the A 260/A 280 values (Table 1).
PCR amplification. 2 μl of genomic DNA diluted 1:10 was used with IllustraTM PuReTaqTM Ready-To-Go PCR Beads (GE Healthcare, Little Chalfont, UK). We used the primers ITS1F (Gardes & Bruns Reference Gardes and Bruns1993), ITS1LM (Myllys et al. Reference Myllys, Lohtander, Källersjö and Tehler1999), ITS4 (White et al. Reference White, Bruns, Lee and Taylor1990) and ITS2KL (Lohtander et al. Reference Lohtander, Myllys, Sundin, Källersjö and Tehler1998) for the nrDNA ITS region, and LR0R (Rehner & Samuels Reference Rehner and Samuels1994) and LR5 (Vilgalys & Hester Reference Vilgalys and Hester1990) for the nrDNA LSU region. The cycle conditions employed were: initial denaturation at 95°C for 3 min, followed by four cycles (95°C for 40 s, 56°C for 40 s, 72°C for 90 s), four cycles (95°C for 40 s, 53°C for 40 s, 72°C for 90 s), 32 cycles (95°C for 40 s, 50°C for 40 s, 72°C for 90 s), and a final extension at 72°C for 6 min. Amplification products were separated by electrophoresis on 1% agarose gels in 1×TAE buffer at 90 V for 20 min, stained with Red SafeTM (iNtRON Biotechnology, Seoul, Korea) and visualized under UV light.
Sequencing. The PCR products were purified by IllustraTM ExoProStarTM 1-step (GE Healthcare, Little Chalfont, UK). One strand was sequenced using the ABI Prism Dye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Foster City, CA, USA) with the forward primers used in the amplification step (ITS1F / ITS1LM and LR0R) at the Unidad de Genómica (Parque Científico de Madrid). Sequence fragments obtained were checked and manually adjusted in BioEdit v7.0.5 (Hall Reference Hall1999).
Overall, we obtained 10 ITS and six nrLSU sequences in our evaluation of the method, that is 45·45% and 42·86% success, respectively, for each marker (Table 1).
The concentration of total genomic DNA was between 3–165 ng/μl; lower values of the range corresponded to the lichenicolous fungi when only very small quantities of minute hyphae were used as starting material, and higher values corresponded to the lichen fungus where a piece of a young lobe was used. The purity of the extracted DNA was not optimal (A 260/A 280<1·8) but that was not indicative of the success in amplification achieved for the selected DNA regions. The particularly low purity of the DNA extracts from Xanthoparmelia specimens (0·76–0·82) is indicative of the presence of co-extracted impurities which may act as PCR inhibitors (possibly extrolites and polysaccharides). When 1/10 dilution of some extracts was evaluated, optimal purity was reached for Lichenostigma cosmopolites 1, Parmelia saxatilis, Parmelina tiliacea, and P. quercina (see Table 1). The dilution of the stock evidently decreases the concentration of any impurities present and better results in the amplification of DNA are obtained.
PCR products and the complete ITS sequences were successfully obtained from the fresh specimens of lichen-forming fungi analyzed (100% success). For older specimens, it was found to be better to use internal primers and amplify shorter regions.
Only a fraction of the lichenicolous fungi amplified correctly, with the resulting sequences from those samples corresponding to the host; a common situation for fungi intimately connected with their hosts.
Good results were acquired for Abrothallus specimens; sequences of at least one analyzed marker were successfully obtained for one, two, and six year-old samples. Clean sequences of both markers were obtained from a fresh specimen of Homostegia piggotii. All attempts to amplify six year-old samples of H. piggotii failed. Although the purity of the extracted DNA was low (0·98–1·27), degraded DNA might also explain the failure and internal primers should then be used. We were not able to amplify any fresh sample of Lichenostigma cosmopolites due to the persistent amplification of its host. In this case, our best results came from older material (11 years old) when there seemed to be less chance of the host lichen DNA being amplified. Specific primers could improve the chances of obtaining correct sequences from fresh material.
We found the Chelex 100 method as used here to be simple and rapid, involving just three steps and taking less than 30 minutes. No organic solvents were used, reducing potential environmental pollution and harm to the operator. The reactive material employed is much cheaper than that commonly used in other methods in lichenology (e.g. CTAB, commercial kits). Furthermore, the DNA extracted using Chelex 100 was of sufficient quality for PCR, especially when fresh material was studied. In addition, the method is suitable for minute samples and when large numbers of samples have to be processed, as relatively few manipulations are required. Moreover, Turan et al. (Reference Turan, Nanni, Brunelli and Collina2015) obtained DNA of better quality from conidia of Venturia using the Chelex-based protocol in comparison to the CTAB protocol. The Chelex-based method also has advantages over direct PCR as a stock of extracted DNA is obtained. That DNA can be stored for future studies either in the freezer in water, or on Whatman® FTA® Cards at room temperature (Gueidan et al. Reference Gueidan, Aptroot, Cáceres and Binh2016). The DNA stock was also used for PCR six months after its extraction when identical results were obtained in Lichenostigma cosmopolites (data not shown). Further improvements to the method to increase the purity of the DNA are being tested, particularly purification by ion exchange resins prior to PCR or Proteinase K treatment of the extracts; these will be reported separately.
Although this technique has been known for almost 30 years (Singer-Sam et al. Reference Singer-Sam, Tanguay and Riggs1989), is commonly used in human forensics (Walsh et al. Reference Walsh, Metzger and Higuchi1991) and has also been successfully used in other eukaryotes (e.g. Pedersen et al. Reference Pedersen, Russell, Newton and Ansell2006; HwangBo et al. Reference HwangBo, Son, Lee, Min, Ko, Liu and Jeong2010), this study appears to be the first time it has been reported for use in lichen-forming and lichenicolous fungi.
In conclusion, we recommend the technique for the routine extraction of any fresh and/or minute material as it provides a reliable, fast and low-cost alternative to the DNA extraction protocols generally used in lichenology today.
This contribution was completed while ZF was in receipt of funding from the Spanish Ministerio de Economía y Competitividad project CGL2014-55542-P, and DLH was on the staff of the Universidad Complutense de Madrid. Our colleague C. Ruibal is gratefully acknowledged for fruitful discussions on the topic. We thank CAI de Genómica y Proteómica del Parque Científico de Madrid for their services. Two anonymous reviewers are acknowledged for their valuable comments on the manuscript.

