INTRODUCTION
Thalassiosira pseudonana is a widely distributed centric (radially symmetrical) diatom (Lowe & Busch, Reference Lowe and Busch1975; Belcher & Swale, Reference Belcher and Swale1977; Hasle, Reference Hasle1978; Ake-Castillo et al., Reference Ake-Castillo, Hernandez-Becerril and Meave Del Castillo1999). It was the first diatom to have its whole genome sequenced (Armbrust et al., Reference Armbrust, Berges, Bowler, Green, Martinez, Putnam, Zhou, Allen, Apt, Bechner, Brzezinski, Chaal, Chiovitti, Davis, Demarest, Detter, Glavina, Goodstein, Hadi, Hellsten, Hildebrand, Jenkins, Jurka, Kapitonov, Kröger, Lau, Lane, Larimer, Lippmeier, Lucas, Medina, Montsant, Obornik, Parker, Palenik, Pazour, Richardson, Rynearson, Valentin, Vardi, Wilkerson and Rokhsar2004), and since then the number of studies on this taxon have increased greatly, resulting in it being widely adopted as a model organism (e.g. Tonon et al., Reference Tonon, Harvey, Qing, Li, Larson and Graham2004; Alverson et al., Reference Alverson, Jansen and Theriot2007, Reference Alverson, Beszteri, Julius and Theriot2011; Hildebrand et al., Reference Hildebrand, Frigeri and Davis2007). These studies have an absolute requirement for phenotypically and genotypically characterized algal strains; however, the strains of this organism deposited in culture collections have not been systematically characterized and compared. For a range of algae, intraspecific characterization has proved invaluable, defining taxa and revealing cryptic diversity amongst strains available in the collections (Müller et al., Reference Müller, Friedl, Hepperle, Lorenz and Day2005; De Martino et al., Reference De Martino, Meichenin, Shi, Pan and Bowler2007; Veselá et al., Reference Veselá, Neustupa, Pichrtová and Poulícková2009), and providing researchers worldwide with an overview of the characteristics of the available strains of a particular species.
Historically diatom taxonomy was primarily based on light microscope observations of the morphology of their cell wall (frustule). Subsequently, the use of electron microscopy resulted in a significant advance for taxonomy, since it revealed additional characters not observable by light microscopy, and facilitated the description of many new diatom genera (see Round et al., Reference Round, Crawford and Mann1990 for an overview). More recently, advances in molecular techniques have demonstrated that diatom diversity is even greater than originally thought (Mann et al., Reference Mann, Chepurnov and Idei2003; Amato et al., Reference Amato, Kooistra, Levialdi Ghiron, Mann, Pröschold and Montresor2007). These techniques have revealed that many species, whose descriptions were based on morphological characters, were actually comprised of a number of cryptic species, with identical or almost identical morphologies (Amato et al., Reference Amato, Kooistra, Levialdi Ghiron, Mann, Pröschold and Montresor2007; Mann et al., Reference Mann, Thomas and Evans2008). Diatom cell division usually (Rose & Cox, Reference Rose and Cox2013) involves a reduction in average cell size often with a change in cell proportions, but can also in many long-term cultured isolates translate into a total loss of shape and morphological characters over time (Round et al., Reference Round, Crawford and Mann1990), making phenotypic identification difficult or even impossible. It is evident that morphologically based studies are not always able to resolve species identities, but a combined approach using molecular techniques can reveal cryptic speciation (Sarno et al., Reference Sarno, Kooistra, Medlin, Percopo and Zingone2005; Amato et al., Reference Amato, Kooistra, Levialdi Ghiron, Mann, Pröschold and Montresor2007; Evans et al., Reference Evans, Wortley, Simpson, Chepurnov and Mann2008; Kooistra et al., Reference Kooistra, Sarno, Balzano, Gu, Andersen and Zingone2008), and in some cases may demonstrate biogeographic patterns (Casteleyn et al., Reference Casteleyn, Chepurnov, Leliaert, Mann, Bates, Lundholm, Rhodes, Sabbe and Vyverman2008; Kooistra et al., Reference Kooistra, Sarno, Balzano, Gu, Andersen and Zingone2008).
A variety of molecular markers and house-keeping genes, such as the gene cytochrome c oxidase (cox1), large subunit of RUBISCO (rbcL), small subunit of the ribosomal DNA (SSU) and the internal transcribed spacer regions (ITS1+ITS2), have been used as ‘barcodes’ to distinguish between diatom species (Evans et al., Reference Evans, Wortley and Mann2007; Moniz & Kaczmarska, Reference Moniz and Kaczmarska2010; Hamsher et al., Reference Hamsher, Evans, Mann, Poulícková and Saunders2011). Additionally, the use of DNA fingerprinting techniques that produce multi-locus banding patterns have been employed to characterize geographic isolates and closely related species (John et al., Reference John, Groben, Beszteri and Medlin2004). Such fingerprinting techniques include Randomly Amplified Polymorphic DNA (RAPD) (Williams et al., Reference Williams, Kubelik, Livak, Rafalski and Tingey1990; Barker et al., Reference Barker, Green, Hayes and Medlin1994; Baillie et al., Reference Baillie, Belda-Baillie and Maruyama2000), Amplified Fragment Length Polymorphism (AFLP) (Vos et al., Reference Vos, Hogers, Bleeker, Reijans, Van De Lee, Hornes, Frijters, Pot, Peleman, Kuiper and Zabeau1995; John et al., Reference John, Groben, Beszteri and Medlin2004; Müller et al., Reference Müller, Friedl, Hepperle, Lorenz and Day2005; De Martino et al., Reference De Martino, Meichenin, Shi, Pan and Bowler2007) and microsatellites (Rynearson & Armbrust, Reference Rynearson and Armbrust2000; Evans et al., Reference Evans, Chepurnov, Sluiman, Thomas, Spears and Mann2009). In particular, AFLP detects DNA polymorphisms in different genomic regions without a requirement for previous sequence knowledge of the organism, it is highly sensitive and with methodological care, reproducible (Meudt & Clarke, Reference Meudt and Clarke2007).
While T. pseudonana is used as one of the principal marine diatom models, a recent study has revealed that the likely ancestor was a freshwater species (Alverson et al., Reference Alverson, Beszteri, Julius and Theriot2011) and that it may subsequently have transitioned to live in brackish to marine waters. The proposal is that T. pseudonana would be better known by its original name, Cyclotella nana Hustedt. This clearly brings into question the relevance of T. pseudonana as a marine diatom model, and potentially might indicate the presence of cryptic diversity amongst strains of this diatom species if this transition from freshwater to marine occurred at different times and/or locations. Moreover, the a priori assumption that a taxon with wide biogeographic distribution would most likely have some level of detectable (cryptic) diversity led us to hypothesize the potential for cryptic speciation within T. pseudonana.
In the present study, a polyphasic approach employing both phenotypic and genotypic characters was used to investigate whether there were identifiable differences among T. pseudonana isolates (including the strain T. pseudonana CCMP1335, which was the first diatom to have its whole genome sequenced) obtained from international Biological Resource Centres (Table 1). The morphology of the strains was investigated using scanning electron microscopy; for the genotypic characterization, proposed DNA barcoding genes were used, as well as a whole genomic approach, AFLP. AFLP analyses are generated for many loci across the whole genome (increasing the possibility of finding polymorphisms), but the technique is highly sensitive to DNA quality (Vos et al., Reference Vos, Hogers, Bleeker, Reijans, Van De Lee, Hornes, Frijters, Pot, Peleman, Kuiper and Zabeau1995). To address this, as outlined by Müller et al. (Reference Müller, Friedl, Hepperle, Lorenz and Day2005), we performed two DNA extractions on different days and three AFLP reactions using both DNA extractions to test the reproducibility of the method. The overall aim was to investigate the morphological and genetic diversity of this species.
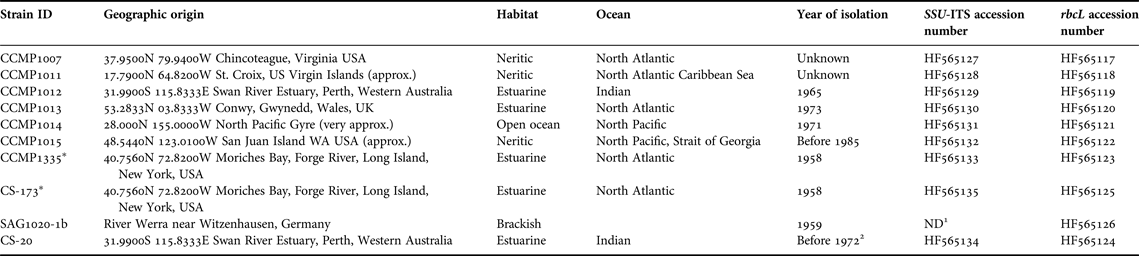
Table 1. Thalassiosira pseudonana strains studied, with strains ID, geographic origin, year of isolation and GenBank accession numbers.

Information from: www.epsag.uni-goettingen.de, ccmp.bigelow.org and www.csiro.au/places/Australian-National-Algae-Culture-Collection
1 Not determined due to poor sequence quality.
2 Ian Jameson (ANACC) personal communication.
* Duplicate strains.
MATERIALS AND METHODS
Strains and culture conditions
Ten T. pseudonana strains were provided by three culture collections, the Provasoli-Guillard National Center for Marine Algae and Microbiota (NCMA, formerly CCMP), the Australian National Algae Culture Collection (ANACC) and the Culture Collection of Algae at Göttingen University (SAG), (for further details see Table 1). For the study, two duplicate strains were included, CCMP1335 and CS-173; the latter was obtained from the ANACC and is a duplicate of CCMP1335, which has been maintained in the ANACC collection since 1984. The comparison of these daughter strains, maintained in two different collections for decades, sought to reveal whether different culture regimes over a time-frame of 2–3 decades can influence phenotypic and/or genetic traits.
All the strains were grown in 100 ml Erlenmeyer flasks containing 50 ml sterile F/2 medium (Guillard, Reference Guillard, Smith and Chanley1975) plus sodium metasilicate, referred to here as F/2+Si (33–34 ppt salinity). The culture conditions were standardized, incubated at 15°C under a 12/12 h light/dark regime (irradiance: ~30 μmol. photons m−2 s−1). All the strains were tested for axenicity on medium containing nutrient agar.
Light and scanning electron microscopy
Cells were harvested in stationary phase; a small volume of each strain was removed for light microscopy and 5 ml used to prepare specimens for SEM.
Live cells were examined using a Polyvar (Vienna, Austria) light microscope and photographs were taken using a Leica DFC320 camera and IM50 (Leica Microsystems, Heerbrugg, Switzerland) imaging systems.
The samples for electron microscopy were acid-washed following the method of Lundholm et al. (Reference Lundholm, Daugbjerg and Moestrup2002), in order to remove all the organic material from the cells. Cleaned diatom frustules were concentrated on 13 mm cover glasses (Menzel-glaser), previously mounted on SEM aluminium stubs (Agar Scientific Ltd, Essex, UK). The stubs were coated using gold palladium; applied at a thickness of 5 nm using a Cressington 208HR sputter-coater and measured using a quartz crystal thickness monitor. Samples were examined with a Zeiss Ultra Plus Field Emission SEM (Carl Zeiss Ltd, UK) (NHM, London, UK) and digital photographs taken for measurements.
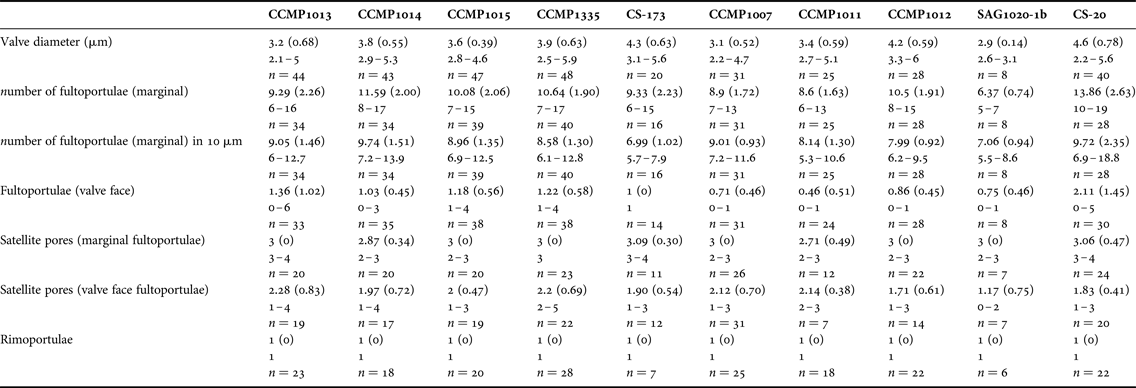
Valve diameter, number and position of fultoportulae (SPs), number and position of rimoportulae (LPS), number of satellite pores in SPs and areola shape and patterns were determined for at least 30 valves using SEM images from each of the 10 T. pseudonana cultured isolates.
DNA extraction, PCR and DNA sequencing
Two millilitres of each T. pseudonana strain were centrifuged at 3800 g in a SIGMA 1–14 microcentrifuge (Sigma-Aldrich Ltd., Dorset, UK) to harvest sufficient cells for extraction. The cells were frozen in liquid nitrogen, ground with a mortar and pestle and the frozen powder transferred to a microcentrifuge tube containing Plant DNAZOL Reagent (Gibco BRL, Grand Island, NY, USA) (approximately 0.3 ml Plant DNAZOL for 0.1 g of plant tissue). Genomic DNA was extracted using the protocol provided by the manufacturer. For AFLP analyses, genomic DNA was extracted twice for each strain on different days in order to identify any potential variation in AFLP patterns due to differences in the extraction process (Müller et al., Reference Müller, Friedl, Hepperle, Lorenz and Day2005).
The SSU and ITS rDNA were amplified according to Luo et al. (Reference Luo, Pflugmacher, Pröschold, Walz and Krienitz2006) from the extracted genomic DNA using the Taq PCR Mastermix Kit (Qiagen, Hilden, Germany) employing the primers EAF3 and ITS055R listed in Table 2.
Table 2. Oligonucleotide primers used to amplify and sequence SSU, ITS, rbcL and CoxI fragments from Thalassiosira pseudonana.
In addition to the nuclear markers, the plastid gene that encodes the large subunit of RuBisCO, rbcL, was sequenced. DNA was amplified using the Taq PCR Mastermix Kit (Qiagen, Hilden, Germany) with the primers DPrbcL1 and DPrbcL7 (Table 2). The PCR conditions were an initial denaturing phase for 3 min (94°C), followed by 30 cycles of 94°C for 10 s, 50°C for 1 min and 68°C for 3 min, with a final extension of 68°C for 7 min.
Two strategies were used to amplify the mitochondrial marker cox1. The first involved amplifying the genomic DNA using the Taq PCR Mastermix Kit (Qiagen, Hilden, Germany) with the primers GAZF2 (Saunders, Reference Saunders2005) and KEdtmR (Evans et al., Reference Evans, Wortley and Mann2007), (Table 2). The PCR protocol comprised an initial denaturation step at 94°C for 4 min followed by 12 touch-down cycles involving: denaturation at 94°C for 30 s, annealing at 65–54°C for 1 min and extension at 72°C for 1 min; followed by another 25 cycles of denaturation at 94°C for 30 s, annealing at 53°C for 1 min and extension at 72°C for 1 min, with a final extension at 72°C for 10 min. The second strategy involved using Takara Ex Taq (Takara Bio Inc., Shiga, Japan) using the PCR conditions described above. The PCR product was then cloned using the TOPO TA Cloning Kit (Invitrogen) following the manufacturer's instructions.
All the PCR products were visualized by agarose gel electrophoresis and purified with the QIAquick PCR Purification Kit (Qiagen, Hilden, Germany) prior to being sent to the GenePool Sequencing Facility (University of Edinburgh, UK) for DNA sequencing using an ABI 3730 (Applied Biosystems) capillary sequencer. The sequences generated were assembled and manually aligned using the sequence editor MacVector 8.1 (Accelrys Inc.). To determine which evolutionary model best fitted the analyses, the values of 56 different models were estimated using the program Modeltest 3.7 (Posada & Crandall, Reference Posada and Crandall1998; Posada & Buckley, Reference Posada and Buckley2004). Phylogenetic trees were created using maximum likelihood method (ML), employing the Tamura–Nei model (Tamura & Nei, Reference Tamura and Nei1993); chosen as the best model according to the Akaike Information Criterion (by Modeltest) via the PAUP 4.0b10 program (Swofford, Reference Swofford2002). To test the confidence of the tree topology, bootstrap analyses were calculated by maximum likelihood (ML, 100 replicates) criteria.
AFLP
Genomic DNA from six axenic strains was extracted using Plant DNAzol as described above. The purity of DNA was estimated using a spectrophotometer (NanoPhotometer 2.0, Implen GmbH, Schatzbogen 52, Germany) and only samples with ratio A 260/A 280≥1.8 were submitted for AFLP analysis. Restriction and ligation of the genomic DNA was performed in a single reaction (Mannschreck et al., Reference Mannschreck, Klemp, Kley, Friedrich, Kühlwein, Wickert, Habram, Matuska and Slemr2002) using the endonucleases MseI and EcoRI (New England Biolabs, Frankfurt, Germany).
Aliquots (5.5 μl) of genomic DNA were incubated with 1 U MseI, 5 U EcoRI and 1 Weiss Unit T4 DNA ligase (New England Biolabs, Frankfurt, Germany) in T4 Ligase buffer with 55 mM NaCl, 0.55 μg BSA, EcoR1 and Mse1 adapters (5 and 50 pmol, sequences in Vos et al., Reference Vos, Hogers, Bleeker, Reijans, Van De Lee, Hornes, Frijters, Pot, Peleman, Kuiper and Zabeau1995) as described in Müller et al. (Reference Müller, Friedl, Hepperle, Lorenz and Day2005). The pre-selective amplification was modified as follows: 4 μl of the reactions were used with primers MseI+0 (GATGAGTCCTGAGTAA) and EcoRI+0 (GACTGCGTACCAATTC) (2.5 pmol each, Vos et al., Reference Vos, Hogers, Bleeker, Reijans, Van De Lee, Hornes, Frijters, Pot, Peleman, Kuiper and Zabeau1995), 10 μl of Taq PCR Mastermix (Qiagen, Hilden, Germany) and 5 μl double distilled water, in a total volume of 20 μl. The parameters for the amplification were 5 min at 94°C, followed by 20 cycles of 20 s at 94°C, 30 s at 56°C and 120 s at 72°C. The quality of the pre-selective amplification was checked on a 1.5% agarose gel, and depending on the amount of generated PCR products observed, these were then diluted (typically between 0 to 1/10) prior to the selective amplification, as described in Müller et al. (Reference Müller, Friedl, Hepperle, Lorenz and Day2005). In the next amplification, 4 μl of the diluted pre-selective amplification products were used in a total reaction volume of 20 μl with the following primer combinations: EcoRI+A (GACTGCGTACCAATTCA, 10 pmol), EcoRI+C (GACTGCGTACCAATTCC, 5 pmol) and MseI+C (GATGAGTCCTGAGTAAC, 10 pmol). EcoRI+A was labelled with the fluorochrome 6-FAM and EcoRI+C with VIC (Applied Biosystems, Foster City, CA, USA) and the selective amplification conditions were 5 min at 94°C, followed by 10 cycles of 20 s at 94°C, 30 s at 65°C with 1°C decrease after each cycle down to 56°C and 120 s at 72°C, followed by 20 cycles of 20 s at 94°C, 30 s at 56°C and 120 s at 72°C. The AFLP reaction was completed twice with the first DNA extraction and for a third time with the second DNA extraction on different days to test the reproducibility of the technique.
To visualize the AFLP, an Agilent 2100 bioanalyser (Agilent Technologies Deutschland, Waldbronn, Germany) was used to size and quantify the AFLP fragments based on capillary gel electrophoresis principles. DNA 12000 LabChips (Agilent Technologies) were prepared and loaded with samples as recommended by the manufacturer and then inserted into the Agilent 2100 bioanalyser for analysis using the Agilent 2100 expert software. The bioanalyser displayed data as computer-generated virtual gels. To analyse the data, the image files produced by the bioanalyser were imported into GelQuest (SequentiX – Digital DNA processing, Klein Raden, Germany) and detection and sizing of the fragments performed. The resulting binary matrix was then exported in NEXUS format and analysed with PAUP 4.0b10 (Swofford, Reference Swofford2002). A distance matrix was constructed using the restriction-site distance of Nei & Li (Reference Nei and Li1979) and this was submitted for neighbour-joining (NJ) analysis. Confidence for the tree topology was tested using bootstrap analyses (2000 replicates). Further AFLP analysis of the same samples was conducted using an ABI 3730 capillary sequencer. A small volume of the selective amplification (1 μl) of the 10-times diluted selective amplification was combined with 1 μl of the 50-times diluted GeneScan 1200 Liz Size Standard (20–1200 bp range; Applied Biosystems, Foster City, CA, USA) and analysed using ABI GeneMapper (GenePool Sequencing Facility, Edinburgh, UK). Electropherograms were imported into and analysed using the GelQuest software (SequentiX – Digital DNA processing, Klein Raden, Germany). For each strain, AFLP fragments were only scored when they were present in at least two of the three replicates over a threshold of 50 relative fluorescent units for automated evaluation, as described by Müller et al. (Reference Müller, Friedl, Hepperle, Lorenz and Day2005). Further manual analysis was undertaken to avoid exclusion of low intensity peaks, and also to avoid inclusion of ‘false’ peaks due to a strong signal in a neighbouring channel or to background noise. The resulting binary matrix was exported in NEXUS format and analysed with PAUP 4.0b10 (Swofford, Reference Swofford2002) as described above.
RESULTS
Morphological characterization
Due to its small size and weakly silicified valves, relatively few morphological features are discernible in T. pseudonana with light microscopy. The cultured strains occurred as single cells and colonies were never observed. Valves were circular, 2–7 μm in diameter (Table 3) containing 4 small chloroplasts.
Table 3. Main morphometric characters observed in Thalassiosira pseudonana strains under study.
Values given for mean (standard deviation in parentheses), followed by range and number of measurements.
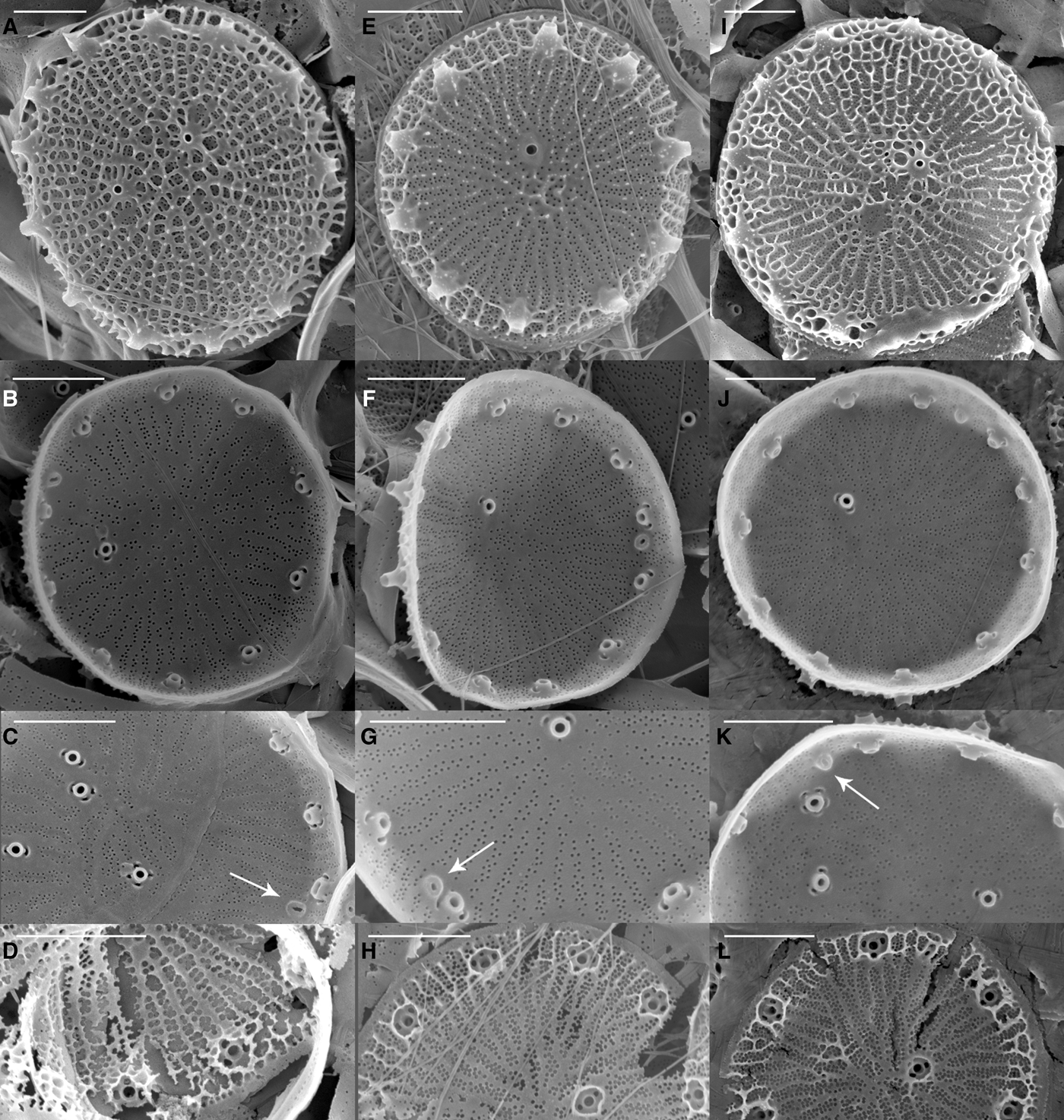
Scanning electron microscopy (SEM) revealed that all the strains were weakly silicified (e.g. Figure 1). Whole cells were disc-like, with flat valve faces and a short pervalvar axis, less than the cell diameter. The valve surface was perforated by radially arranged rows of small pores, the pattern being disrupted by the valve face fultoportulae (strutted processes, SPs) (Figure 1). Most specimens had irregular radial ribs, crossed by tangential ribs forming polygonal, or elongated, areolae on the external valve surface (e.g. Figure 1). There were 0–6 valve face SPs with 1–4 satellite pores, and 6–19 marginal SPs, each with 2–5 satellite pores (Table 3). The number of marginal SPs appeared to be correlated with the cell diameter (Table 3). In all valves examined one circular to oval rimoportulae (labiate process, LP) was observed, situated between 2 marginal SPs. The LP was usually midway between the SPs, but sometimes closer to one of them. External valve views revealed that both valve face and marginal SPs were surrounded at their bases by an external siliceous collar and that the marginal SPs had tube-like openings, with the external tube being longer than the internal one (e.g. Figure 1F). SEM observations revealed that there was as much (or more) variation in morphometric characters among specimens within a strain as among strains, precluding the use of any of these characters to distinguish among the strains (Table 3). There was also considerable variation in the degree of areola development and valve silicification between specimens and strains under the same culture conditions and stage of growth (Figure 1).
Fig. 1. Scanning electron micrographs of three Thalassiosira pseudonana strains from different geographic origins: (A–D) T. pseudonana CCMP1013 (Wales, UK); (E–H) T. pseudonana CCMP1014 (North Pacific gyre); (I–L) T. pseudonana CCMP1335 (Long Island, New York, USA). The first row shows external valve views, the second row shows internal views, the third row shows details of the rimoportula (arrow) and the fourth row shows valves in formation (broken during SEM preparation). Scale bars equals 1 μm.
Genotypic characterization
DNA BARCODING
Pairwise comparison of nuclear (SSU and ITS rDNA) and plastid (rbcL) nucleotide sequences revealed no variable sequence positions among the strains examined. Mitochondrial cox1 gene sequences were not amplifiable by PCR using the primer pairs described (Table 2).
AFLP
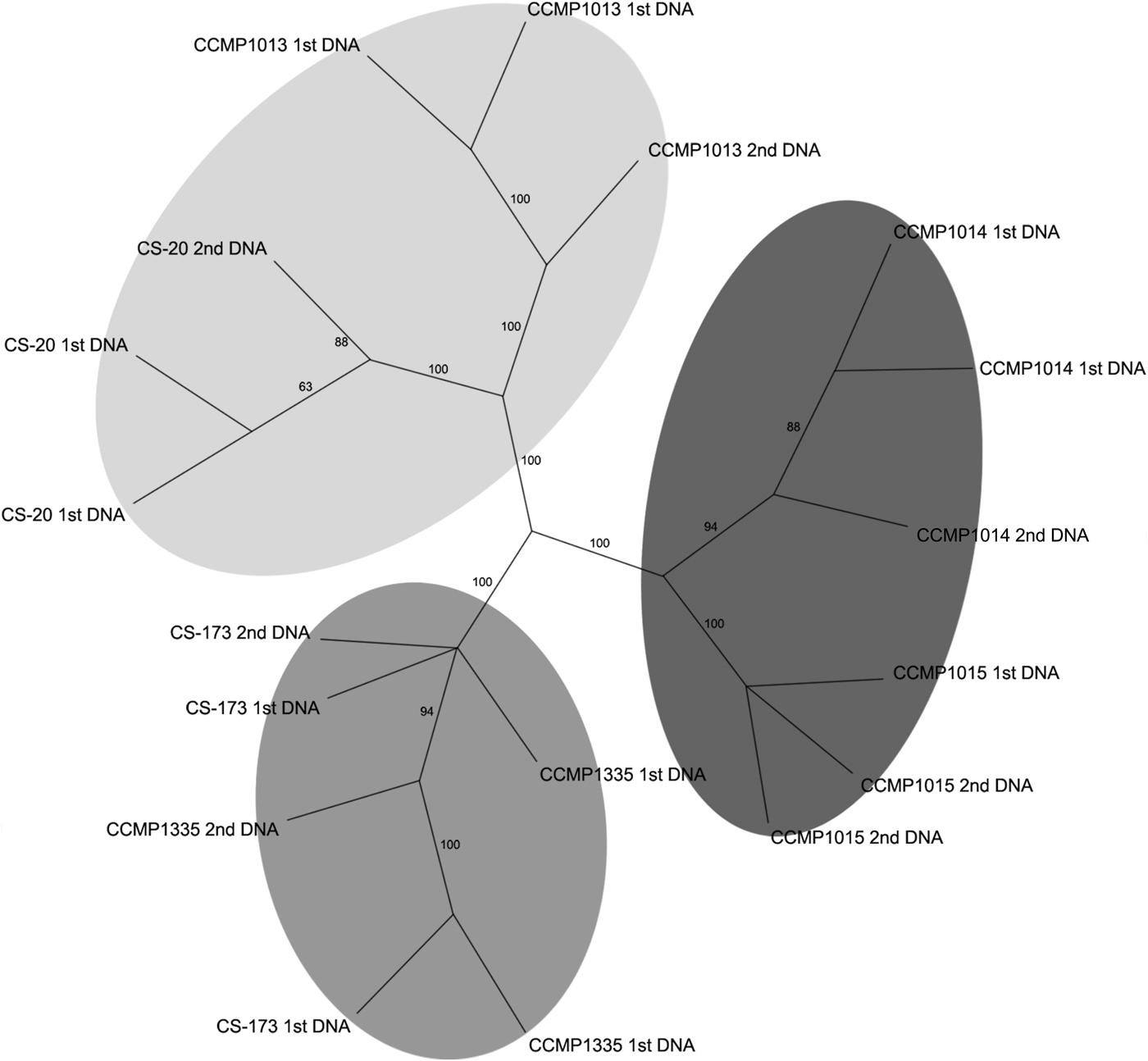
An initial assessment of genetic variation among the axenic strains involved analysis of the products obtained from the selective amplification on an Agilent 2100 bioanalyser. The virtual gel generated was imported into GelQuest (SequentiX – Digital DNA processing, Klein Raden, Germany) and 108 fragments were scored, and the resulting binary matrix was exported for phylogenetic analyses. The unrooted NJ tree revealed that the strains studied subdivided into three supported lineages (Appendix 1). Subsequently automated evaluation of AFLP data using the ABI 3730 capillary sequencer revealed high variation among strains due to differences in the intensity of some fragments. Those fragments with fluorescence intensity lower than a pre-set threshold value were not detected by automated evaluation, resulting in false-negative variation in the AFLP pattern. To resolve these issues it was necessary to manually inspect the AFLP electropherograms to reduce the number of artifactual differences among strains and replicates. After the manual refinement, a total of 547 fragments were recognized and the resulting binary matrix used for subsequent phylogenetic analyses. The resulting NJ analysis demonstrated that the strains subdivided into the same three well-supported lineages (Figure 2) obtained with the bioanalyser (Appendix 1). This three-way division in the populations was the best supported of several hypothetical groupings tested using Analysis of molecular variance (AMOVA) (Appendix 2). However, this AMOVA analysis was not significant (P = 0.06), probably due to the low levels of diversity and small number of samples.
Fig. 2. Neighbour-joining tree (1000 replicates) based on AFLPs from automated evaluation with manual refinement.
DISCUSSION
Polyphasic taxonomic characterization
In the present study, a polyphasic approach combining morphological and genotypic techniques was used to determine whether diversity exists among a range of T. pseudonana isolates available from algal culture collections in the UK, USA, Germany and Australia. Due to diatom size reduction and the possible loss of defining morphological characters over generations of laboratory culture (Warren et al., Reference Warren, Day, Brown, Hurst, Knudsen, McInerney, Stezenbach and Walter1997), polyphasic taxonomic approaches have a greater capacity to identify or distinguish between microalgae at both species and strain level than purely morphological approaches (Pröschold et al., Reference Pröschold, Marin, Schlosser and Melkonian2001; Sarno et al., Reference Sarno, Kooistra, Medlin, Percopo and Zingone2005; Kooistra et al., Reference Kooistra, Sarno, Balzano, Gu, Andersen and Zingone2008). This approach has been successfully employed by Sarno et al. (Reference Sarno, Kooistra, Medlin, Percopo and Zingone2005) to investigate the diversity in the genus Skeletonema, revealing four new species, and Beszteri et al. (Reference Beszteri, John and Medlin2007) who demonstrated that Cyclotella meneghiniana Kützing comprises a species complex of reproductively isolated taxa and was not a monophyletic group, as originally proposed.
MORPHOLOGICAL CHARACTERIZATION
While light microscopy is useful for the morphological-based identification of algae based on discriminating features such as size, shape, process number and distribution, the small size and weakly silicified frustules of T. pseudonana required electron microscopy to resolve the finer, distinguishing morphological features. Scanning electron microscopy (SEM) observations of the main frustule features confirmed Hasle & Heimdal's (Reference Hasle and Heimdal1970) findings: specimens were 4–9 μm in diameter, weakly silicified; most of the specimens had a central rosette close to the central process; some specimens had irregular radial ribs with no areolae; others had polygonal areolae in the valve centre, elongated at the middle and equilateral polygonal areolae in the marginal zone; other specimens had well-developed areolae over the entire valve surface. The number of marginal processes (corresponding to fultoportulae) was 8–17, with a rectangular or oval slit (corresponding to rimoportulae) at about the same distance from the valve margin as the marginal processes.
Alverson et al. (Reference Alverson, Beszteri, Julius and Theriot2011) examined three T. pseudonana strains, including the fully sequenced strain T. pseudonana CCMP1335, and compared their observations with Cyclotella nana isolectotype. SEM observations in the present study largely confirmed the observations of Alverson et al. (Reference Alverson, Beszteri, Julius and Theriot2011) and the characters reported for the C. nana isolectotype; however, they reported there were three satellite pores per marginal strutted process for all the strains studied. In the present study we observed three satellite pores in T. pseudonana CCMP1335; however, the number of satellite pores per marginal strutted process varied from 2 to 4 for the other strains studied including the daughter strain CS-173. This variation could potentially be related to culture conditions, although in this study all the strains were cultivated under the same conditions. The authors speculate that the variation was due to inherent morphological plasticity in cultures, as the variation was observed among and within strains.
Previous studies examining intraspecific variability and biogeographic distribution of diatoms have reported that the combination of a few morphological characters was sufficient to distinguish between strains, for example, Pseudo-nitzschia pungens (Grunow ex Cleve) Hasle (Casteleyn et al., Reference Casteleyn, Chepurnov, Leliaert, Mann, Bates, Lundholm, Rhodes, Sabbe and Vyverman2008). De Martino et al. (Reference De Martino, Meichenin, Shi, Pan and Bowler2007) have previously demonstrated very high intraspecific morphological variability within Phaeodactylum tricornutum Bohlin that was easily recognizable by light microscopy, although the correlation between morphology, biogeographic distribution and phylogeny was limited. However, in the present study the morphological range of the 10 T. pseudonana strains was so similar that no distinguishing morphometric features were identified (Table 3, Figure 1). In fact, there was a greater morphometric variation within an individual strain than among strains.
DNA BARCODING
The pairwise distance matrix derived from the nuclear (ITS and SSU) and plastid (rbcL) genes of T. pseudonana demonstrated that, for these genes, the strains shared identical nucleotide sequences. Clearly, while barcoding has previously been successful in identifying cryptic species in other groups (Darienko et al., Reference Darienko, Gustavs, Mudimu, Rad-Menendez, Schumann, Karsten, Friedl and Pröschold2010; Stern et al., Reference Stern, Andersen, Jameson, Küpper, Coffroth, Vaulot, Le Gall, Véron, Brand, Skelton, Kasai, Lilly and Keeling2012), and in other diatom genera (Alverson et al., Reference Alverson, Jansen and Theriot2007; Evans et al., Reference Evans, Wortley and Mann2007; Moniz & Kaczmarska, Reference Moniz and Kaczmarska2009, Reference Moniz and Kaczmarska2010), the absence of any nucleotide variation among the T. pseudonana strains indicates that for T. pseudonana these genes have reached their resolution limit at the species level. The outcome of this evidence would suggest that there is no cryptic diversity within this model diatom, based on the limited number of available culture collection strains.
No nucleotide sequence data was obtained from the mitochondrial gene (cox1), although two primer sets were evaluated. Evans et al. (Reference Evans, Wortley and Mann2007) have suggested that additional PCR primers need to be developed for a universal cox1 diatom barcoding system to be viable. This suggests low universality of this mitochondrial marker amongst diatoms, possibly due to the presence of introns (Armbrust et al., Reference Armbrust, Berges, Bowler, Green, Martinez, Putnam, Zhou, Allen, Apt, Bechner, Brzezinski, Chaal, Chiovitti, Davis, Demarest, Detter, Glavina, Goodstein, Hadi, Hellsten, Hildebrand, Jenkins, Jurka, Kapitonov, Kröger, Lau, Lane, Larimer, Lippmeier, Lucas, Medina, Montsant, Obornik, Parker, Palenik, Pazour, Richardson, Rynearson, Valentin, Vardi, Wilkerson and Rokhsar2004), and that the mitochondrial gene may not be the best choice for barcoding diatoms, as previously noted by Hamsher et al. (Reference Hamsher, Evans, Mann, Poulícková and Saunders2011).
Overall, the absence of any identifiable differences in the morphology and DNA barcoding sequences of the different T. pseudonana cultures leads to the conclusion that all these strains belong to a single species. Moreover, this species is apparently cosmopolitan, based on the isolation origins of the strains from both hemispheres and different oceanic basins (Table 1). From the point of view that T. pseudonana is a model diatom species, this study provides evidence that studying any of these strains will apply as a study of a single unified species. However, we considered the lack of any differences to be unusual, as other studies of diatoms have readily observed differences (Alverson et al., Reference Alverson, Jansen and Theriot2007; Evans et al., Reference Evans, Wortley and Mann2007; Moniz & Kaczmarska, Reference Moniz and Kaczmarska2009, Reference Moniz and Kaczmarska2010), and therefore, if the study of T. pseudonana as a model diatom should have ecological relevance, further study is needed to establish whether the culture collection strains analysed here are representative of T. pseudonana in the field.
AFLP intraspecies characterization
The use of AFLP analyses has been able to resolve genetic variations when other genetic markers have reached their resolution limits and to reveal previously undetected genetic diversity and differentiate among strains within populations (Werner et al., Reference Werner, Olschewski and Mergenhagen2001; John et al., Reference John, Groben, Beszteri and Medlin2004; Müller et al., Reference Müller, Friedl, Hepperle, Lorenz and Day2005). The unexpected finding that there were no morphological or genotypic differences to differentiate among the T. pseudonana strains led us to use AFLP to explore intraspecific diversity among six axenic isolates. The two daughter strains (T. pseudonana CCMP1335 and T. pseudonana CS-173), which were maintained independently for a period of >20 years, were included to test whether variations in culture/ maintenance regimes could affect genetic stability.
The analysis resolved three clusters of strains, supported by bootstrap analysis (Figure 2). One cluster was composed of two oceanic strains (CCMP1014 and 1015) and the other two clusters contained estuarine strains CS-173 and CCMP1335, and CCMP1013 and CS-20 respectively. With the availability of further axenic strains, it may be possible using AFLP or more recent techniques such as RAD sequencing (Davey et al., Reference Davey, Hohenlohe, Etter, Boone, Catchen and Blaxter2011), or even whole genome sequence analysis to further elucidate the drivers underpinning the genetic diversity evident in the different strains.
DUPLICATED CLONE
Two strains derived from a single clone and maintained in separate culture collections for >20 years were included in the AFLP analyses to examine whether extended laboratory cultivation had resulted in any ‘genotypic drift’. AFLP analysis demonstrated that both strains of the single clone formed a discrete and supported cluster, which in fact contained greater genetic variation between DNA extraction replicates than among the strains (Figure 2). Thus, it is concluded that within the resolution limits of this technique 26 years of separate cultivation had no detectable effect on the genotype.
Very low clonal diversity was surprising since, speculatively, genomic variation could be expected among strains of T. pseudonana due to its wide geographic distribution and its capacity to grow under a range of ecological conditions. Casteleyn et al. (Reference Casteleyn, Chepurnov, Leliaert, Mann, Bates, Lundholm, Rhodes, Sabbe and Vyverman2008) previously reported genetic diversity among different geographic isolates of P. pungens using the ITS1–5.8S–ITS2 rDNA region; however, in our study the ITS region lacked the resolution to distinguish among T. pseudonana strains. In a similar study using analyses of the rDNA operon, John et al. (Reference John, Groben, Beszteri and Medlin2004) revealed geographic clades within the Alexandrium tamarense species complex, although further AFLP analyses demonstrated very high genetic diversity even within single populations. In the present study, AFLPs showed apparently low genetic diversity among T. pseudonana strains, despite the taxon's putative transition from freshwater to marine environments (Alverson et al., Reference Alverson, Beszteri, Julius and Theriot2011), and its wide distribution.
Since T. pseudonana is widely distributed around the globe, is able to grow in a range of ecological niches, and has a putative ancestral freshwater origin (Alverson et al., Reference Alverson, Beszteri, Julius and Theriot2011), we had hypothesized that the culture collection strains would show evidence of cryptic diversity, and potentially the presence of different species. The evidence presented in this study shows instead that T. pseudonana appears to represent a single diatom species, displaying very low clonal diversity, with no morphological features that would reliably discriminate among strains, identical DNA barcodes and a potentially low degree of genomic diversity based on AFLP analyses.
ACKNOWLEDGEMENTS
The authors thank the Provasoli-Guillard National Center for Marine Algae and Microbiota (NCMA, formerly CCMP), the Australian National Algae Culture Collection (ANACC, formerly CSIRO) and the Culture Collection of Algae at Göttingen University (SAG) for providing cultures and kindly sharing information about the strains. We also thank Dr Alex Ball for his expertise and advice in the use of the scanning electron microscope, and the Natural History Museum (NHM) for the use of its facilities.
FINANCIAL SUPPORT
This research was supported by the UK Natural Environment Research Council (National Capability funding for CCAP) and NERC Molecular sequencing grant NERC MGF 154.
Supplementary materials and methods
The supplementary material for this article can be found at http://www.journals.cambridge.org/MBI