INTRODUCTION
The autochthonous Mediterranean marine fauna is mostly of Atlantic affinities (Sabelli & Taviani, Reference Sabelli, Taviani, Goffredo and Dubinsky2014), having originated with the re-establishment of the Atlanto-Mediterranean connection (5.33 million years ago (Ma)), after the Messinian Salinity Crisis (from 5.971 to 5.33 Ma: Manzi et al., Reference Manzi, Gennari, Hilgen, Krijgsman, Lugli, Roveri and Sierro2013) had probably nearly exterminated the stenoecious marine biota (Taviani, Reference Taviani2002; Bianchi et al., Reference Bianchi, Morri, Chiantore, Montefalcone, Parravicini, Rovere and Stambler2012). Local alpha diversity, however, is currently under siege by the continuous arrival of alien species (Sabelli & Taviani, Reference Sabelli, Taviani, Goffredo and Dubinsky2014). Allochthonous elements mostly enter the Mediterranean via the Suez Canal, although a conspicuous contribution to the alien component currently comes from human activities (aquaculture, leisure boating, commercial maritime traffic). The early morphological detection of easily-identified shallow water species attracted a number of scientists, becoming one of the main local research fields, especially in the Eastern Mediterranean (Galil & Goren, Reference Galil, Goren, Goffredo and Dubinsky2014 and references therein).
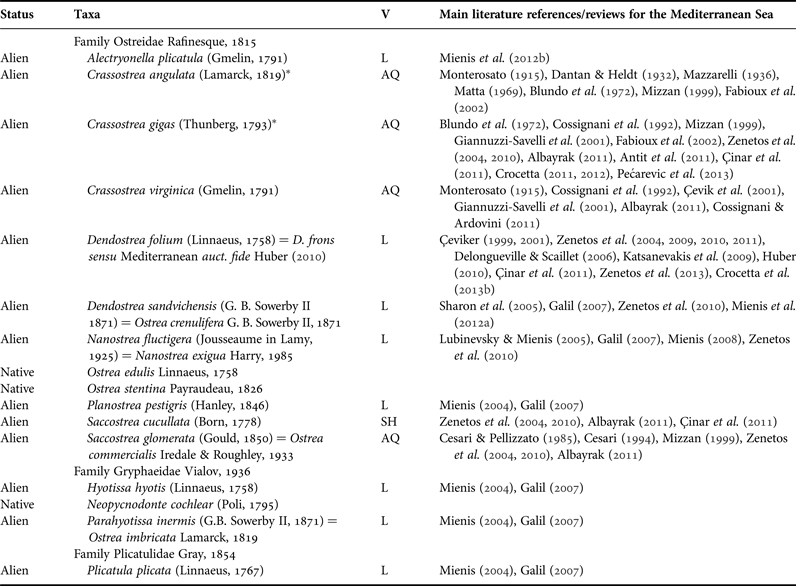
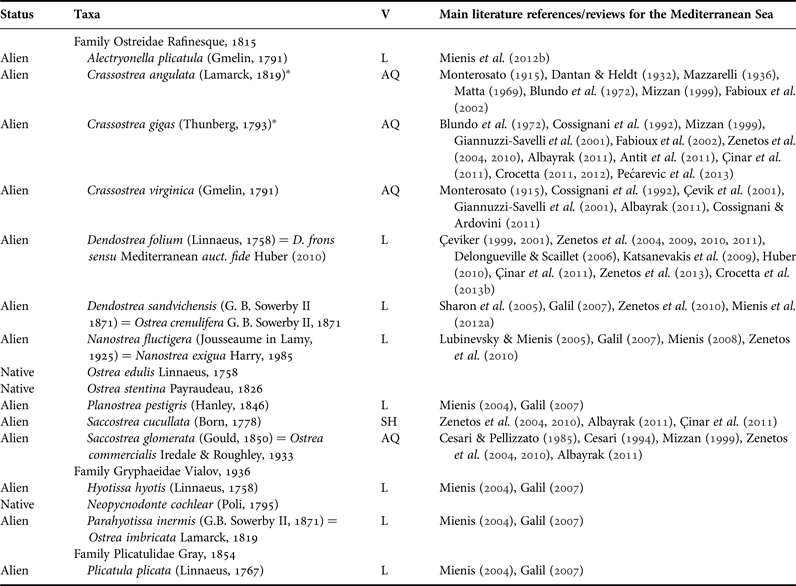
Mollusca currently account for the highest number of known species introduced in the Mediterranean (~200) and, according to literature data, within some families the number of native species is currently outnumbered by that of the alien ones (Zenetos et al., Reference Zenetos, Gofas, Verlaque, Çinar, Garcia Raso, Bianchi, Morri, Azzurro, Bilecenoglu, Froglia, Siokou, Violanti, Sfriso, San Martin, Giangrande, Katağan, Ballesteros, Ramos-Esplà, Mastrototaro, Ocaña, Zingone, Gambi and Streftaris2010, Reference Zenetos, Gofas, Morri, Rosso, Violanti, Garcia Raso, Çinar, Almogi-Labin, Ates, Azzurro, Ballesteros, Bianchi, Bilecenoglu, Gambi, Giangrande, Gravili, Hyams-Kaphzan, Karachle, Katsanevakis, Lipej, Mastrototaro, Mineur, Pancucci-Papadopoulou, Ramos-Esplà, Salas, San Martin, Sfriso, Streftaris and Verlaque2012; Sabelli & Taviani, Reference Sabelli, Taviani, Goffredo and Dubinsky2014). Oysters and relatives, for instance, are represented in the Mediterranean by species belonging to three families: (1) the family Ostreidae Rafinesque, 1815, with two accepted native species, Ostrea edulis Linnaeus, 1758 and Ostrea stentina Payraudeau, 1826, and ~10 alien species reported by several authors during the last century; (2) the family Gryphaeidae Vialov, 1936, with one accepted native species and two Erythraean aliens; and (3) the family Plicatulidae Gray, 1854, with one Erythraean species only (Table 1 and main Mediterranean references therein).
Table 1. Native and alien nominal species of oysters and relatives recorded from the Mediterranean Sea, with plausible/suspected vectors (V, for alien species only) and main references. AQ, aquaculture; SH, shipping; L, lessepsian entry.
*, the taxonomic status of Crassostrea gigas and C. angulata is still controversial, with some authors considering them as different species (e.g. Lapègue et al., Reference Lapègue, Batista, Heurtebise, Yu and Boudry2004; Liu et al., Reference Liu, Li, Kong, Yu and Zheng2011) and others regarding them as conspecific (e.g. Lόpez-Flores et al., Reference Lόpez-Flores, de la Herrán, Garrido-Ramos, Boudry, Ruiz-Rejón and Ruiz-Rejón2004; Reece et al., Reference Reece, Cordes, Stubbs, Hudson and Francis2008). They are here conservatively treated as different species.
However, given the severely misleading morphological patterns in these bivalve families (see discussion in, e.g. Lam & Morton, Reference Lam and Morton2003, Reference Lam and Morton2004; Huber, Reference Huber2010), the need for a genetic confirmation is evident before assessing their presence and distribution in the basin. Nevertheless, studies confirming or elucidating the distribution in the Mediterranean of alien oysters are mostly lacking, with the sole exception of the occurrence in the northern Adriatic Sea of both sibling taxa Crassostrea gigas (Thunberg, 1793) and Crassostrea angulata (Lamarck, 1819) (Fabioux et al., Reference Fabioux, Huvet, Lapègue, Heurtebise and Boudry2002). This has resulted often in chaos in the literature, with authors reporting or deleting species from lists of aliens according to subjective views only, and often without any discussion of the rationale.
Among small oysters (genera Alectryonella and Dendostrea), four nominal species have been cited from the eastern Mediterranean Sea (Table 1). Dendostrea sandvichensis (G. B. Sowerby II, 1871) and Alectryonella plicatula (Gmelin, 1791) have been recorded from Israel only on the basis of the following morphological features: D. sandvichensis (recorded as A. crenulifera) is diagnosed by a cup-shaped lower valve and a flat upper valve, a yellowish-green coloured interior and the exterior of the upper valve of a dirty white colour, often with some weak reddish lines, whilst A. plicatula has a shallow lower valve and a flat upper valve, a yellowish-green internal colour and the upper valve is purple–red externally (Mienis et al., Reference Mienis, Zaslow and Rittner2012b). Dendostrea folium (Linnaeus, 1758) and Dendostrea frons (Linnaeus, 1758) were recorded from Lebanon, Cyprus and Greece without any morphological description (Table 1). Both species are well known to largely overlap in shell morphology, being very variable and showing different ecomorphs, with or without a submedian ridge and a highly variable number of plications, and it is difficult to separate those records from the specimens of D. sandvichensis recorded from Israel. Huber (Reference Huber2010) suggested the exclusion of D. frons (a Caribbean native species: Huber, Reference Huber2010) from the Mediterranean basin on the basis of biogeographical considerations, but his suggestion was not generally accepted, and D. frons has still been reported for some eastern Mediterranean localities (e.g. Zenetos et al., Reference Zenetos, Koutsogiannopoulos, Ovalis and Poursanidis2013) and in general reviews (Nunes et al., Reference Nunes, Katsanevakis, Zenetos and Cardoso2014). However, diagnostic characters differentiating Dendrostrea nominal species are weak, since general outline and shape of valves are known to reflect the nature of the substrate, and the internal and external colour may vary even in the same specimen (e.g. the specimen in Figure 2F, G starts with a red external colouration which then becomes white).
Therefore, a combined morphological–molecular approach may probably help in re-assessing the alpha taxonomy of this group and identifying effective diagnostic characters. To test the reliability of currently available molecular data, samples of small oysters showing extremely different morphological characters and encompassing the known morphological variation (cup-shaped or flat lower valve; flat or rounded upper valve; yellowish-green, whitish or with malachite-green patches internal colour; dark or light reddish, whitish or white with weak reddish lines external colour; presence/absence of regular/irregular rounded ribs; presence/absence of plications at the margins; and massive/scarce presence of chomata: see Figures 2 and 3) were collected at four Mediterranean localities (two in Greece, Astypalia Island and Rhodes Island, and two in Turkey, Kekova and Olympos) spanning most of their range in the Mediterranean. They were then sequenced for the two mitochondrial barcode markers (Liu et al., Reference Liu, Li, Kong, Yu and Zheng2011) with the widest taxonomic coverage in oysters: the 16S rDNA and the COI (GenBank: accessed on 31 January 2014). The nuclear ITS2 rDNA is a further potential candidate barcode marker for bivalves (Salvi et al., Reference Salvi, Bellavia, Cervelli and Mariottini2010; Salvi & Mariottini, Reference Salvi and Mariottini2012), but the coverage of the sequence in GenBank was too low in the small oysters to allow a reliable comparison, and it was thus discarded. The sequences obtained have been compared with those available in GenBank, and a possible identification at the species level in a DNA-barcoding fashion has been finally tested.
MATERIALS AND METHODS
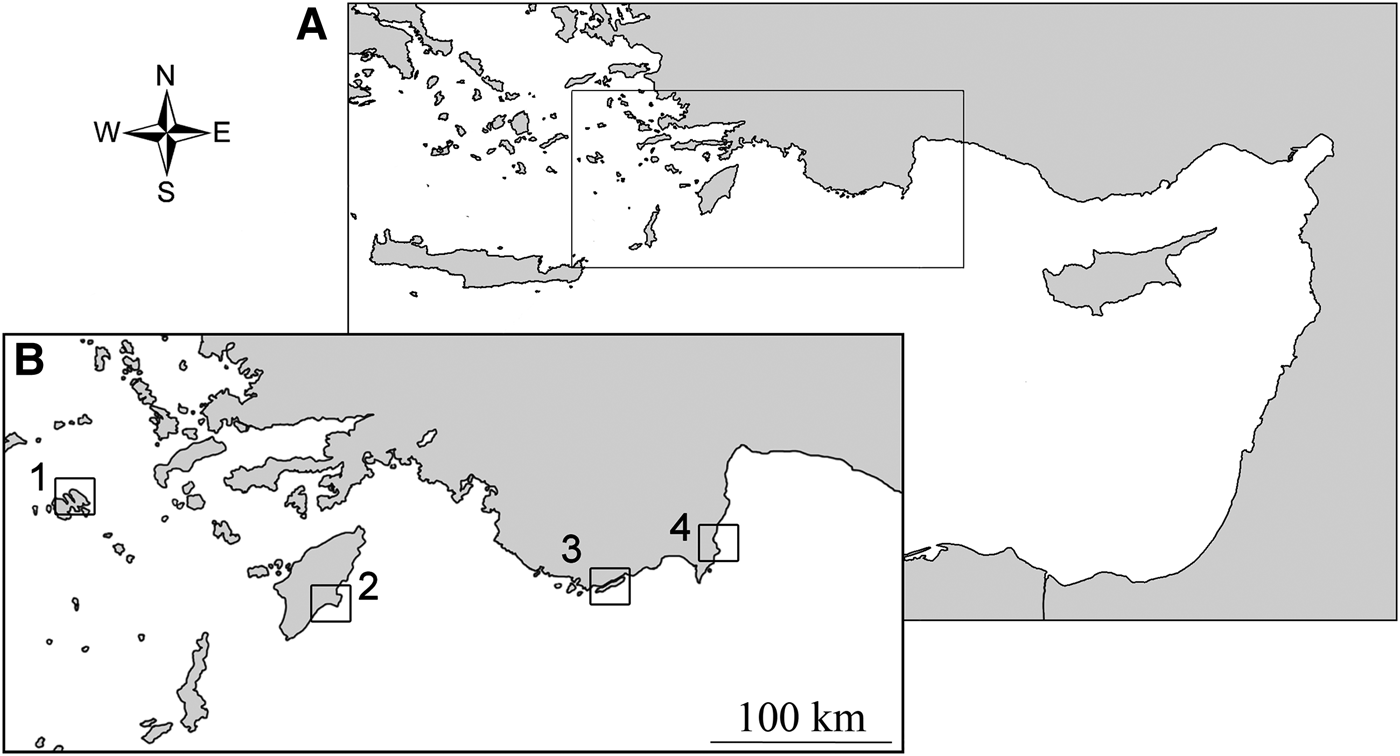
Samples were collected at two Greek ((1) Astypalia (36°35′13″N 026°24′08″E) and (2) Rhodes (36°02′51″N 027°58′93″E)) and two Turkish sites ((3) Kekova (36°10′19″N 029°50′46″E) and (4) Olympos (36°23′40″N 030°28′44″E)), at a depth of 0.1–3 m (Figure 1). At all sites, the oysters were present in high densities of up to 80–100 individuals per square metre. Specimens were collected by hand while diving and fixed in 100% ethanol upon collection. Then, an a priori morphological identification was attempted according to the most recent guides and papers dealing with the involved species (e.g. Sharabati, Reference Sharabati1984; Oliver, Reference Oliver1992; Bosch et al., Reference Bosch, Dance, Moolenbeek and Oliver1995; Sharon et al., Reference Sharon, Benayahu and Mienis2005; Zenetos et al., Reference Zenetos, Konstantinou and Konstantinou2009; Huber, Reference Huber2010; Mienis et al., Reference Mienis, Rittner, Rilov and Almog2012a, Reference Mienis, Zaslow and Rittnerb), but this failed due to the presence of an ambiguous combination of characters typical of distinct species. A piece of tissue was dissected from the foot for DNA extraction, and the remaining tissues were stored, preserved in 100% ethanol with the relevant voucher shell, at the Department of Biology and Biotechnologies, ‘La Sapienza’ University (Figures 2 and 3: voucher ID, BAU1412–BAU1419). Total genomic DNA was extracted using a standard proteinase K phenol/chloroform method with ethanol precipitation, as reported in Oliverio & Mariottini (Reference Oliverio and Mariottini2001). A fragment of the mitochondrial 16S rDNA was amplified by PCR from eight specimens using the universal primers 16Sar-L and 16Sbr-H (Palumbi et al., Reference Palumbi, Martin, Romano, McMillan, Stice and Grabowski2001). The DNA-barcode fragment of the mitochondrial cytochrome oxidase I (COI) was amplified by PCR from the same specimens using the universal primers LCO1490 and HCO2198 (Folmer et al., Reference Folmer, Black, Hoeh, Lutz and Vrijenhoek1994). All amplicons were sequenced by Macrogen Inc. (Seoul, South Korea), using the same PCR primers. Forward and reverse sequences were assembled and edited, and the resulting consensus sequences of each specimen were readily aligned by hand. Accession numbers for the sequences are KJ946438–KJ946453.
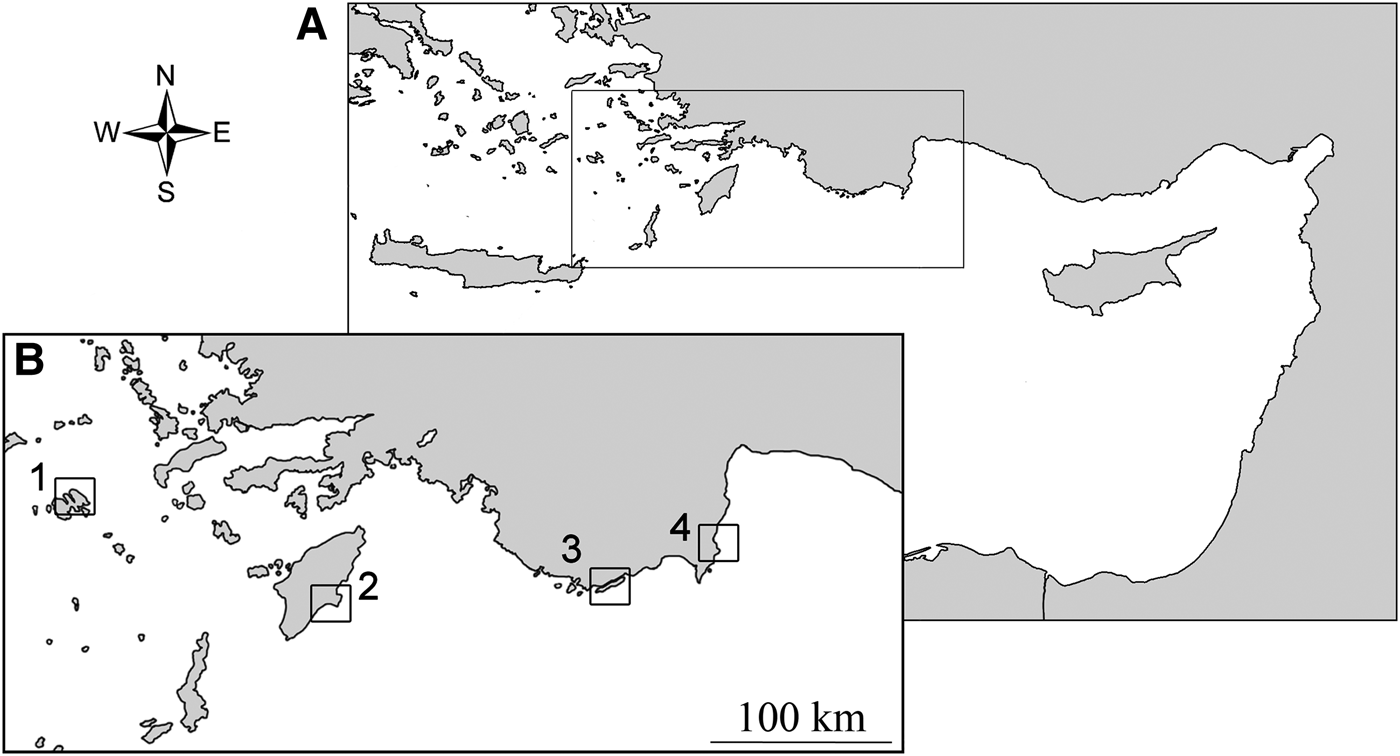
Fig. 1. (A) Location of the sampling area in the eastern Mediterranean Sea; (B) map of the sampling sites: 1, Astypalia; 2, Rhodes; 3, Kekova; 4, Olympos (see Material and Methods for details).
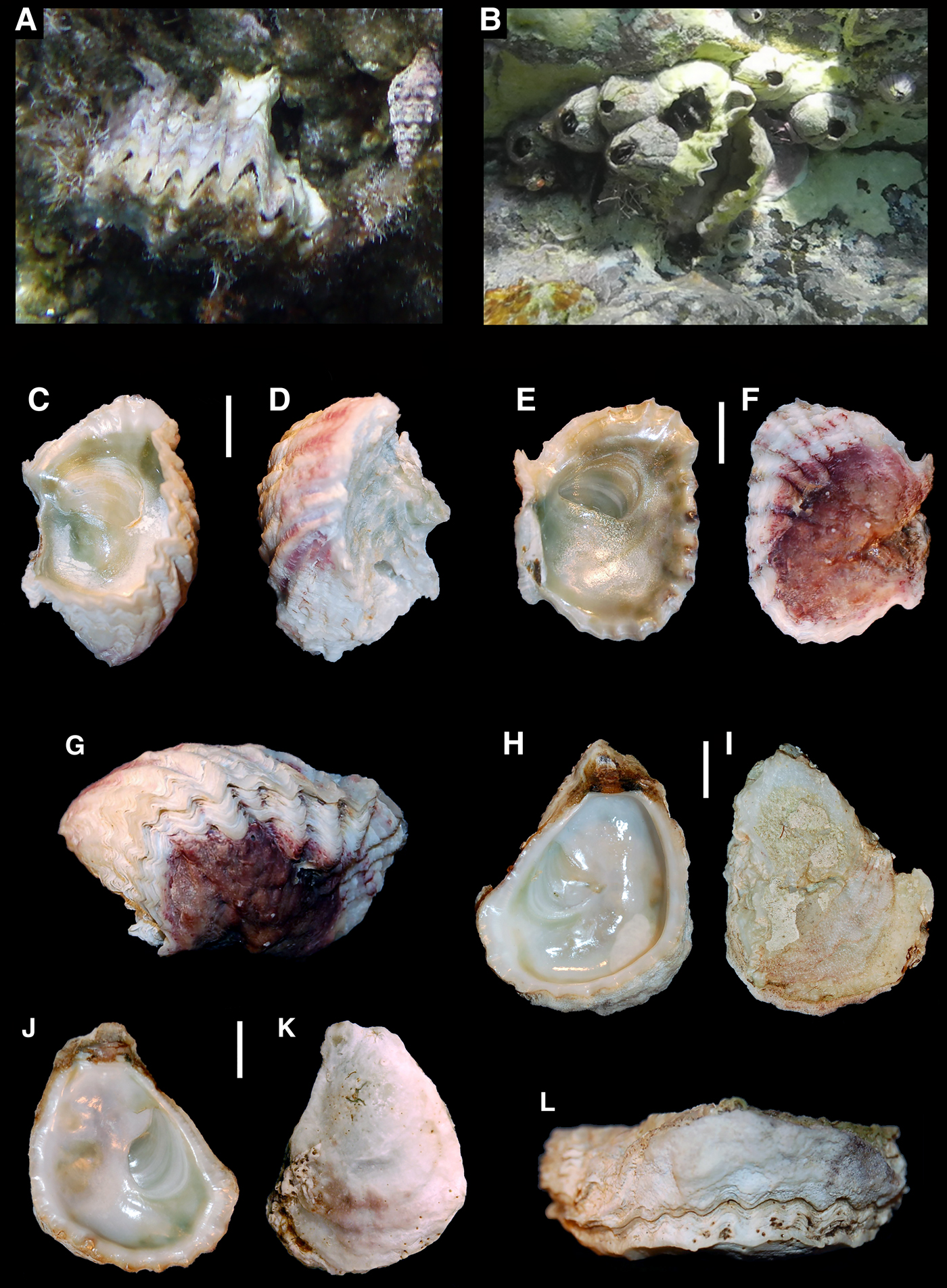
Fig. 2. Small oysters from the Mediterranean Sea: (A) Rhodes (Greece), 50 cm depth, in situ photograph; (B) Astypalia (Greece), 30 cm depth, in situ photograph; (C–G) BAU1412, Rhodes (Greece); (H–L) BAU1413, Rhodes (Greece). Scale bar: 1 cm.
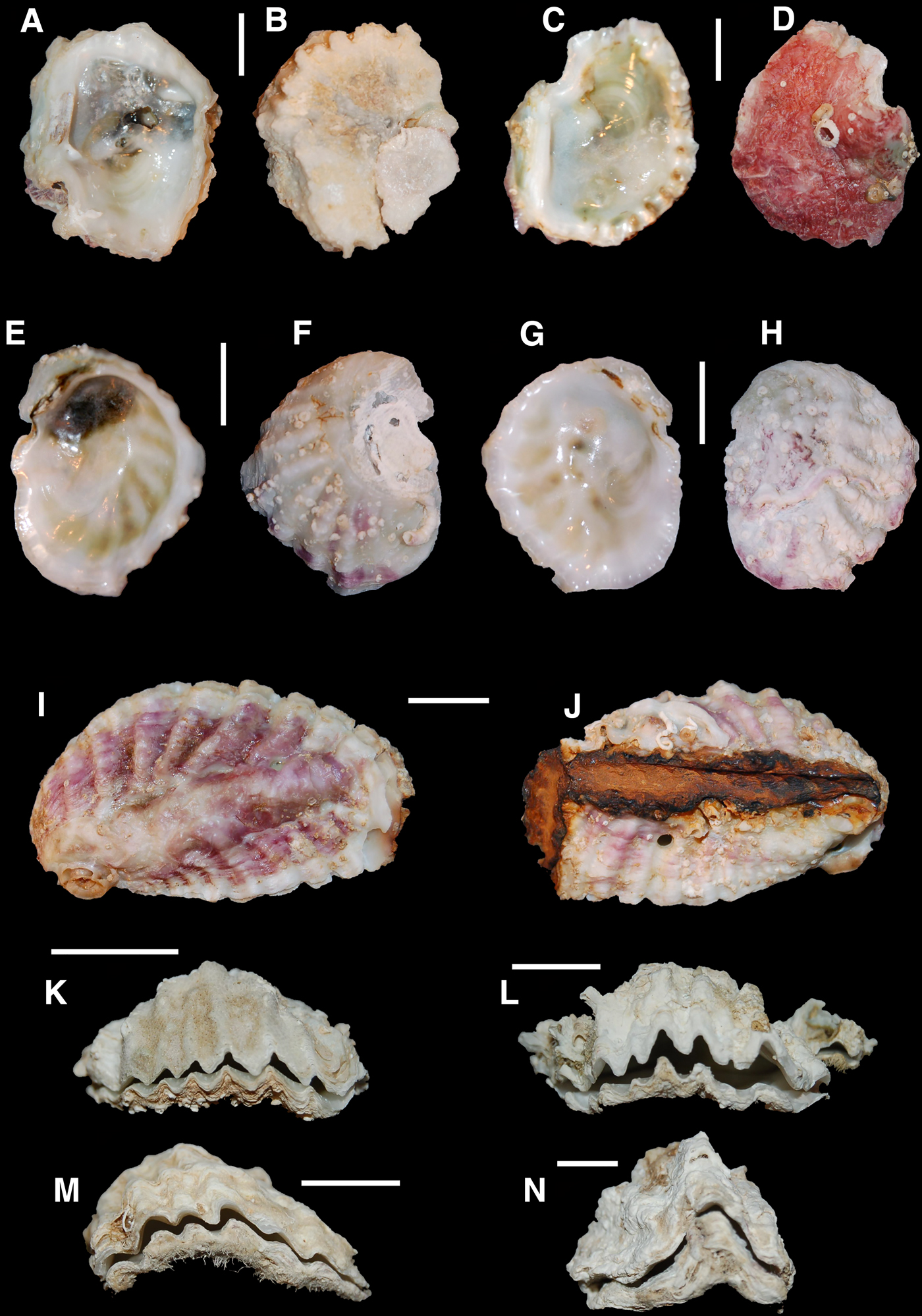
Fig. 3. Small oysters from the Mediterranean Sea: (A–D) BAU1414, Astypalia (Greece); (E–H) BAU1415, Astypalia (Greece); (I, J) Astypalia (Greece), −2 m depth, empty shell from the sampled population; (K) BAU1416, Kekova (Turkey); (L) BAU1417, Kekova (Turkey); (M) BAU1418, Olympos (Turkey); (N) BAU1419, Olympos (Turkey). Scale bar: 1 cm.
A total of 650 (16S) and 923 (COI) ostreid sequences were retrieved from the GenBank, corresponding to specimens identified at species (43 taxa for 16S; 32 taxa for COI) or genus level (24 entities for 16S and 72 entities for COI, determined as ‘sp.’). Of these, 166 (16S) and 145 (COI) sequences were excluded from the database being either too short or too divergent to be reliably included in the analysis. Since our focus was on the lower limit of the interspecific ranges, deleting highly divergent sequences would not bias the analysis. In a first phylogenetic analysis subsets of 92 (16S) and 74 (COI) ostreid sequences, representing all included nominal genera, plus the 16S and COI sequences generated in this study for the small-oysters (N = 8) and three gryphaeid sequences to serve as outgroup (alignments: 16S, 515 bp; COI, 620 bp), were used. Phylogenetic relationships among the sequences were inferred by neighbour-joining (NJ) and maximum likelihood (ML) using the best model of nucleotide substitution, by the software MEGA 5.0 (Tamura et al., Reference Tamura, Peterson, Peterson, Steker, Nei and Kumar2011), bootstrapped over 1000 replicates.
According to the results of these first analyses the retained sequences (484 16S and 782 COI) were pooled into distinct matrices, corresponding to the major clades recovered, each aligned by Clustal_X 1.83 (Thompson et al., Reference Thompson, Gibson, Plewniak, Jeanmougin and Higgins1997), and the final complete alignments were trimmed to the minimum number of corresponding nucleotides (all alignments available from the authors on request). For each gene the three datasets were: a Saccostrea-dataset including all Saccostrea spp. sequences (192 16S sequences, 501 bp; 91 COI sequences, 548 bp); a Crassostrea-datset including all Crassostrea spp. sequences (158 16S sequence, 499 bp; 582 COI sequences, 548 bp); and an Ostreine-dataset including all remaining sequences (134 16S sequences, 497 bp; 109 COI sequences, 548 bp) ascribed to the genera Ostrea, Ostreola and Lopha, and also the so-called small oysters (genera Dendostrea, Alectryonella and allies, including our new sequences). Pooling the sequences in distinct matrices, allowed an easier alignment of the 16S sequences, and, by limiting the need for gaps, allowed retention of longer alignments without having to discard ambiguously aligned partitions. A cumulative matrix including all 484 16S sequences, aligned for 506 bp, required the exclusion of 40 positions of ambiguous alignment.
Analysis of the nucleotide sequences were performed by using MEGA version 5 (Kumar et al., Reference Kumar, Tamura and Nei2004; Tamura et al., Reference Tamura, Peterson, Peterson, Steker, Nei and Kumar2011). The observed ‘p’ and the Kimura's 2-parameter (K2p) (Nei & Kumar, Reference Nei and Kumar2000) pairwise genetic distances were estimated within each dataset.
Updated nomenclature and taxonomy follow the World Register of Marine Species (accessed on 31 January 2014).
RESULTS
Phylogenetic patterns in the Ostreidae
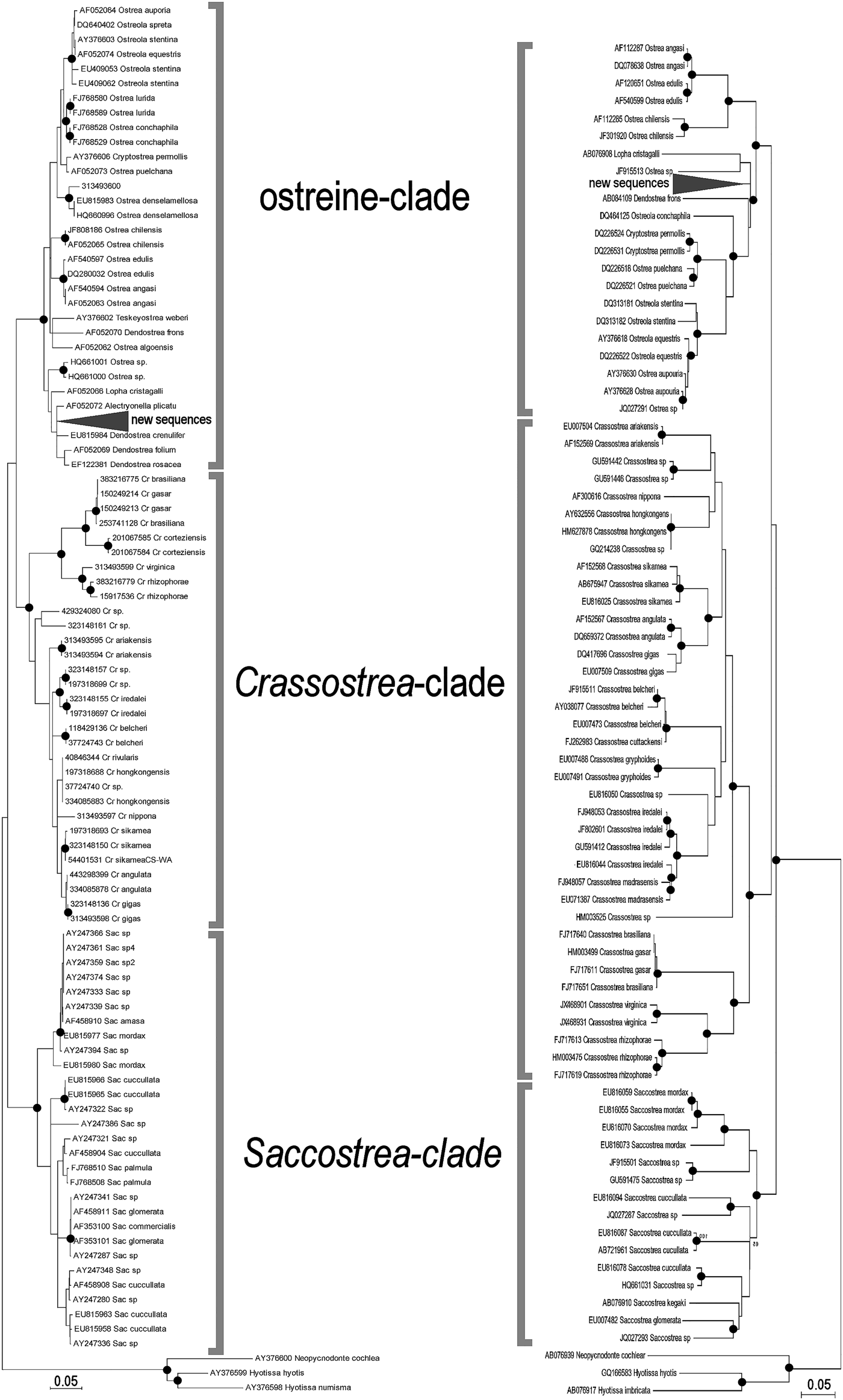
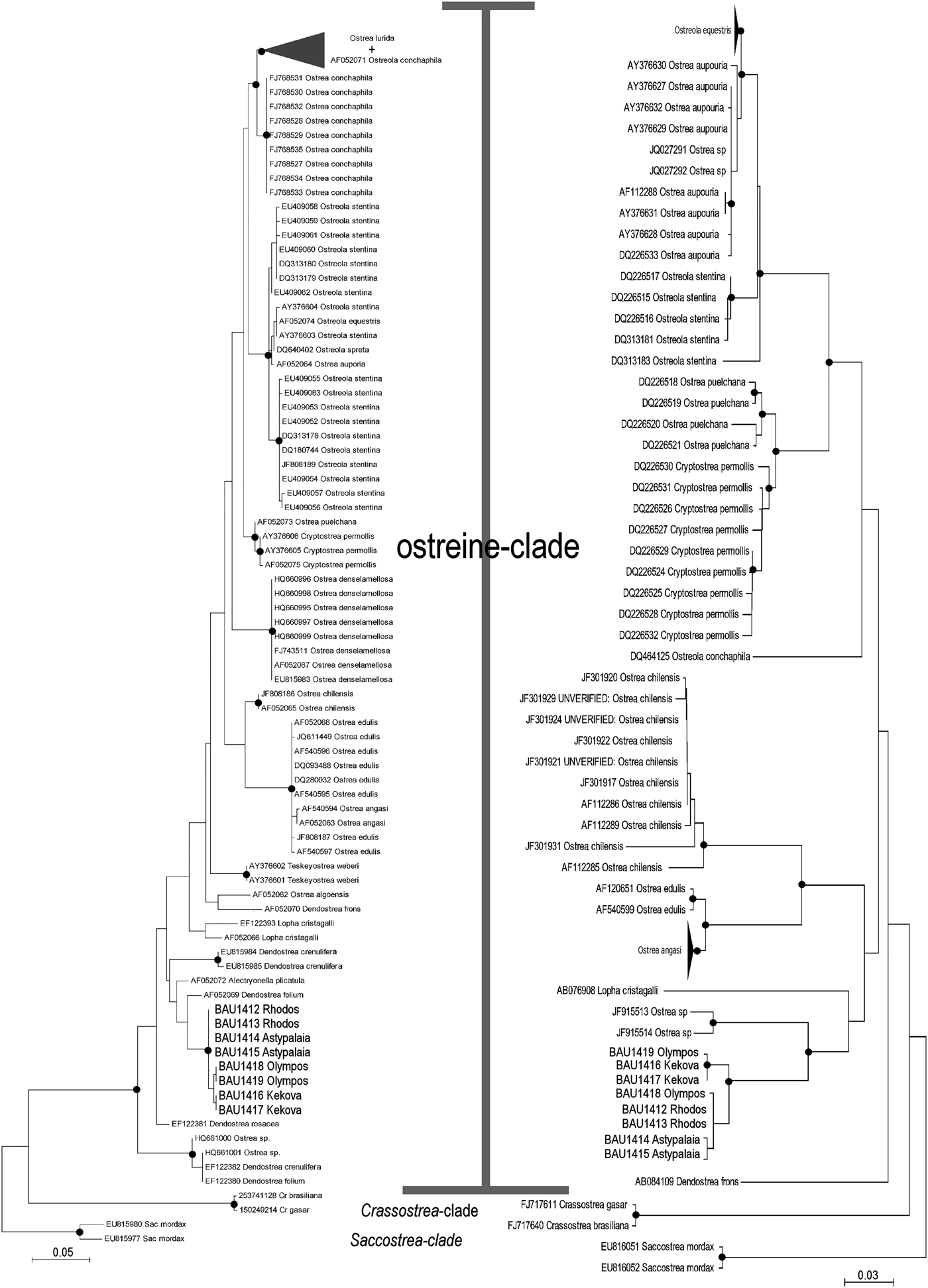
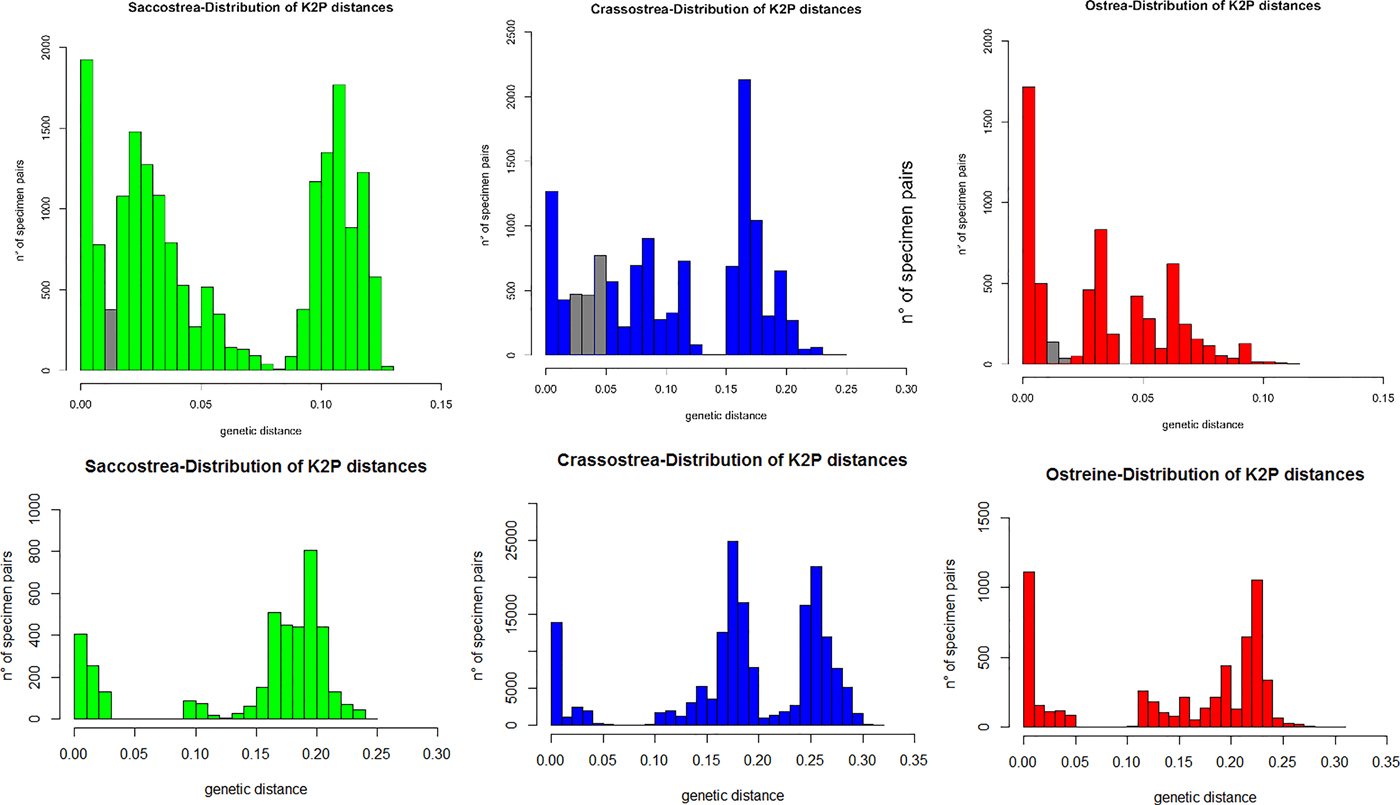
The ML trees recovered by MEGA on the two subsets of 95 (16S) and 74 (COI) ostreid sequences (with three gryphaeids as outgroup), using a GTR (General Time Reversible; Tavaré Reference Tavaré1986) model of evolution, and 1000 bootstrap replicates, are reported in Figure 4 (the topology of the respective NJ trees were almost identical, and only the bootstrap supports at the node are reported). For both datasets the sequences were split into three major clades: a Saccostrea-clade, a Crassostrea-clade, and an Ostreine-clade. The sequences of the small-oysters (Dendostrea-related, including GenBank sequences and our alien Mediterranean samples) were interspersed within the ostreine-clade. Accordingly, ML and NJ analyses (1000 bootstrap) were performed using the Ostreine-dataset with one Crassostrea and one Saccostrea as outgroups (16S: 134 + 2 sequences, alignment 555 bp; COI: 109 sequences, alignment 548 bp) producing the trees in Figure 5. The K2p genetic distances were calculated within each matrix, and the frequency of pairwise distance values among the sequences were plotted in the histograms of Figure 6.
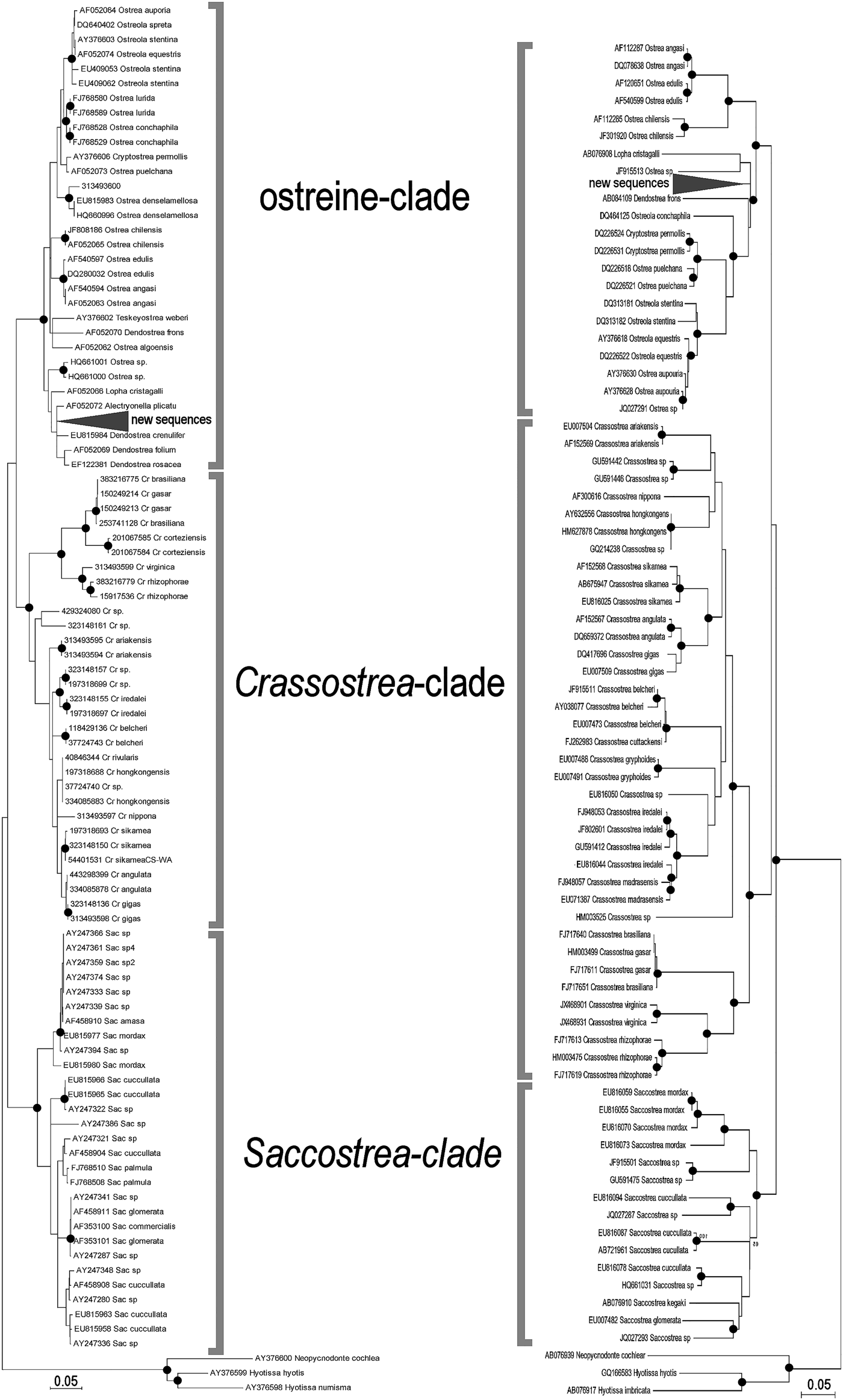
Fig. 4. Maximum likelihood (K2p evolutionary model) phylogenetic trees of selected sequences, encompassing all ostreid genera represented in the GenBank (16S left: log likelihood −3004.1772: COI right: log likelihood −9163.8453. The topologies obtained after NJ and BI analyses recovered the same major clades. Closed circles indicate a bootstrap support (1000 NJ and ML bootstrap replicates) >90% and BI posterior support >95%. Cr, Crassostrea; Sac, Saccostrea.
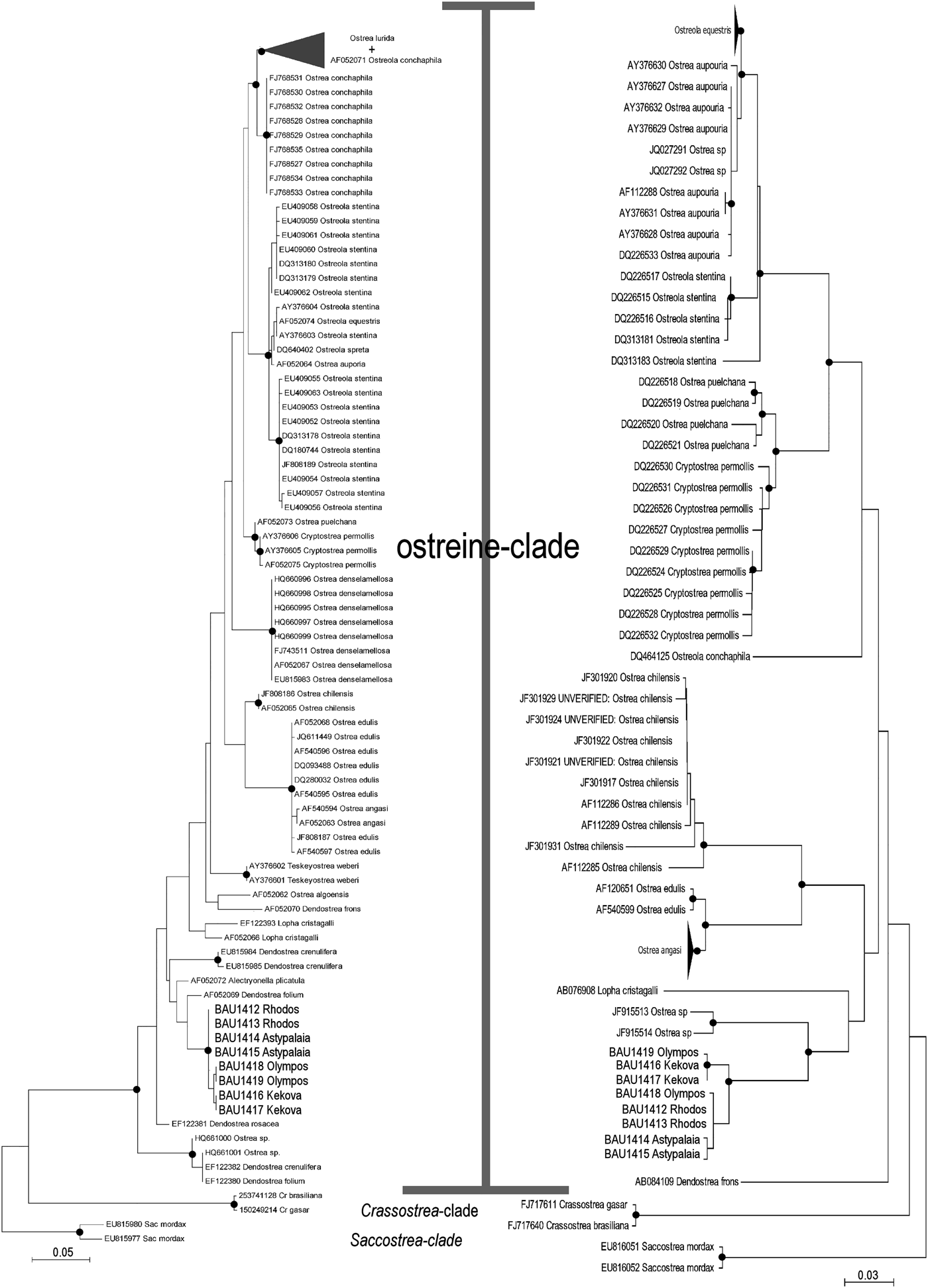
Fig. 5. Maximum likelihood (K2p evolutionary model) phylogenetic trees of the ostreine dataset (16S left: log likelihood −2968.3177: COI right: log likelihood −3634.5067). The topologies obtained after NJ and BI analyses recovered the same major clades. Closed circles indicate a bootstrap support (1000 NJ and ML bootstrap replicates) >90% and BI posterior support >95%. Cr, Crassostrea; Sac, Saccostrea.
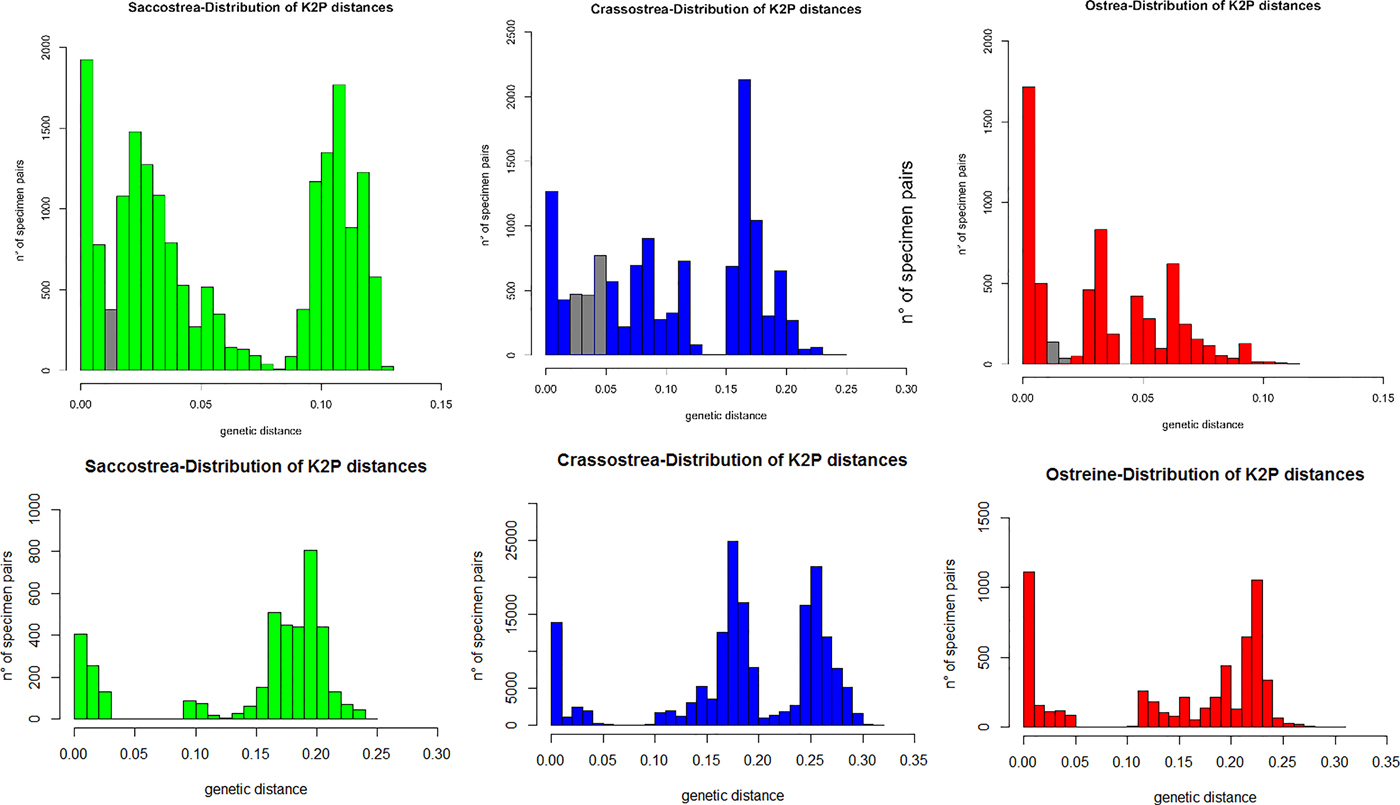
Fig. 6. Histogram representing the frequency of pairwise K2p distance values among 16S (upper row) and COI (lower row) sequences. The grey bars indicate overlaps of presumed intraspecific and interspecific pairwise distances in each dataset.
The visual inspection of the NJ trees (Supplementary Figures S1–S6) derived from each distinct dataset, allowed to identify the clades corresponding to the most probable putative species. According to these patterns, the following maximum intraspecific and minimum interspecific K2p genetic distances were scored in each dataset:
CRASSOSTREA-DATASET
16S
With the exception of a highly diverging sequence from a single specimen ascribed to Crassostrea rhizophorae (AJ312938: with K2p distance 2% from all other C. rhizophorae) all intraspecific distances were <1%. The smallest interspecific distances were 1.2%.
COI
Eight sequences annotated as Crassostrea belcheri (GU591442–GU591447; JF915478, JF9154789) formed a separate clade in the NJ tree, with a minimum K2p divergence of 13.0% from other species, and a K2p divergence of 19.3% from other C. belcheri. The sequence FJ262985 annotated as C. gryphoides was likely a C. belcheri (1.1% K2p divergence from C. belcheri, 17.7%, from other C. gryphoides). The specimen FJ262983 ascribed to C. cuttackensis was likely a C. belcheri (0.55% K2p divergence with other C. belcheri). Very low distance values were obtained between sequences nominally ascribed to distinct species in complexes of doubtful taxonomic status (C. gigas/C. angulata, C. madrasensis/C. iredalei and C. brasiliana/C. gasar), which probably do not include more than one species each. Once accounting for these cases, all intraspecific K2p distances were <4.0% and all interspecific distances were >9.5%.
SACCOSTREA-DATASET
16S
The higher distance values (5%) were observed between specimens of the Saccostrea cucullata/S. palmula complex, where possibly more than one species were involved. The remaining sequences displayed values of distance <2% for presumed intraspecific comparisons, and >3% for interspecific ones.
COI
The sequences annotated as S. cuccullata clustered in three lineages with divergence as high as 19.54%. Sequences identified as S. mordax formed two divergent clades with a minimum genetic distance of 12.05%. These values were similar to the average interspecific divergence between Saccostrea species (16.9%), thus suggesting that both S. cuccullata and S. mordax may represent species complexes. The sequences AY038076 assigned to S. cuccullata was likely from a specimen S. mordax complex. Several sequences identified as Saccostrea sp. have been assigned either to known species (N = 3) or to unidentified lineages (N = 24) which may subtend up to three undescribed species. Once accounting for possible species complexes and undescribed lineages, all the intraspecific distances were <3.37% and all interspecific distances were >12.24%.
OSTREINE-DATASET
16S
Within the ostreine-dataset, the presumed intraspecific distances ranged from 0 to a maximum of 2% (in comparisons among the O. stentina/spreta complex). Among the presumed interspecific comparisons, the lowest values were shown by pairwise distances of O. conchaphila vs O. lurida (1.6–2%; Polson et al. (Reference Polson, Hewson, Eernisse, Baker and Zacherl2009) recently confirmed O. conchaphila as a different species from O. lurida), whilst all other comparisons yielded values >3%. Remarkably, the sequences from specimens of Cryptostrea permollis (AY376605–6, AF052075) resulted virtually identical to those of Ostrea puelchana (AF052073: 0.2–0.7%).
COI
Distance values up to 7.29% were recorded between sequences ascribed to Ostrea chilensis. On the other hand, species pairs such as Ostrea edulis/Ostrea angasi, Ostreola equestris/Ostrea aupouria, and Cryptostrea permollis/Ostrea puelchana showed allegedly interspecific distances lower than 2.81%.
DISCUSSION
A clear threshold between intraspecific and interspecific pairwise comparisons of 16S and COI ostreid sequences did not emerge after the analysis of datasets derived from the GenBank and including our new sequences, although many of the overlaps could be discussed in the framework of taxonomic revisions of the complexes involved (e.g. Crassostrea rhizophorae, Saccostrea cucullata/palmula, Ostrea stentina/spreta and O. conchapila/lurida). However, values higher than 3% for the 16S and of 4% for the COI, seemed to consistently indicate interspecific comparisons, while values lower than 1% (16S) and 10% (COI) were scored in intraspecific comparisons. The sequences from our Mediterranean samples formed highly supported monophyletic groups with both datasets, and pairwise distances ranged from 0 to 0.7% (16S) and from 0 to 0.8% (COI). The COI sequences in the GenBank did not provide any potential conspecific candidate. The closest sequences were JF915513–JF915514, labelled ‘Ostrea sp.’ from Sungai Menghulu (Malaysia), 13–14% divergent from the Mediterranean ones, thus clearly not conspecific. Furthermore, they were not identified at the species level and the phylogenetic pattern clearly indicated that they do not belong in Ostrea but rather are in the Dendostrea clade. All 16S Mediterranean sequences resulted very close to the single sequence of a specimen from Aitutaki (Cook Islands) (AF052069) registered under the name Dendostrea folium by Jozefowicz & Ó Foighil (Reference Jozefowicz and Ó Foighil1998). The locality of this sample is within the range of this taxon (Pacific Ocean: see discussions in Huber, Reference Huber2010). The distance values obtained with the Mediterranean samples (3–3.7%) support a close relationship, yet are not conclusive for testing conspecificity. Furthermore, sequences ascribed to the genus Dendostrea did not form a monophyletic group, nor did sequences ascribed to either D. folium (Linnaeus, 1758) or D. crenulifera (G.B. Sowerby II, 1871) (Figure 5).
Historically, taxonomy of both native and alien Mollusca of the Mediterranean has been almost entirely based on shell characters (e.g. Keller, Reference Keller1883; Pallary, Reference Pallary1912; Haas, Reference Haas1948; Barash & Danin, Reference Barash and Danin1986), that are still commonly used in the early detection and discovery of alien species. Morphological variation of molluscan shells as an adaptive response to environmental pressures, however, has been widely documented (e.g.: Pascoal et al., Reference Pascoal, Carvalho, Creer, Rock, Kawaii, Mendo and Hughes2012; Solas et al., Reference Solas, Sepúlveda and Brante2013). Although the best known taxonomic problems concern the class Gastropoda, oysters (family Ostreidae and allied) represent typical examples of shell morphological plasticity due to environmental pressures, since valve morphology may strongly reflect the nature of the substrate and/or the tidal regime (Gunter Reference Gunter1950; Seilacher et al., Reference Seilacher, Matyla, Wierzbowski, Bayer and Seilacher1985). Morphological characters are often not reliable for differentiating closely related species, and nomenclatural confusion may derive by misidentifications (Korringa, Reference Korringa1952; Boudry et al., Reference Boudry, Heurtebise and Lapègue2003; Huber, Reference Huber2010 and references therein). A combined use of paralleled molecular and morphological markers has become central, in the last decade, to solve issues of oyster identifications and taxonomic relationships (e.g. Lam & Morton Reference Lam and Morton2003, Reference Lam and Morton2004; Lόpez-Flores et al., Reference Lόpez-Flores, de la Herrán, Garrido-Ramos, Boudry, Ruiz-Rejón and Ruiz-Rejón2004, Reference Lόpez-Flores, Ruiz-Rejόn, Cross, Rebordinos, Robles, Navajas-Pérez and de la Herrán2010; Wang et al., Reference Wang, Guo, Zhang and Zhang2004, Reference Wang, Zhang, Liu and Guo2008; Reece et al., Reference Reece, Cordes, Stubbs, Hudson and Francis2008; Liu et al., Reference Liu, Li, Kong, Yu and Zheng2011), further prompting for a wider use of genetic analyses also on alien species in the Mediterranean Sea to delineate dispersal pathways and clarify doubtful identifications (e.g. Albano et al., Reference Albano, Rinaldi, Evangelisti, Kuan and Sabelli2009; Crocetta et al., Reference Crocetta, Macali, Furfaro, Cooke, Villani and Valdés2013a; Valdés et al., Reference Valdés, Alexander, Crocetta, Baki Yokeş, Giacobbe, Poursanidis, Zenetos, Cervera, Caballer, Galil and Schembri2013).
Our GenBank search, however, showed that for oysters the taxonomic coverage of the classical barcode gene (COI) was slightly lower than for 16S. Both markers (COI and 16S) revealed some potential utility for oyster DNA-taxonomy, albeit with some limitation since some non-homogeneous variation seems to exist across the different clades in the family. Nevertheless, the analysis of the pairwise distances in a phylogenetic context allowed the drawing of some conclusion and raised several concerns. The possibility of using a DNA-barcoding approach for oysters mostly relies on the availability of a baseline of pedigreed sequences in the GenBank (i.e. sequences from figured specimens properly identified by specialists, better if from type localities of the nominal taxa); this is still lacking, at least for the species here investigated. Furthermore, a major problem with the use of DNA sequences in identifying small oysters is that several COI and 16S sequences in the GenBank originated from specimens not properly identified. With the exception of Dendostrea frons, with a sequence largely diverging from the other small oysters, the remaining sequences ascribed to the genus Dendostrea show some close relationships, but do not form a holophyletic group, nor do sequences ascribed to either D. folium or D. crenulifera. Given the importance of oysters from many points of view, and the frequent translocation of specimens resulting in alien populations of oysters in many seas, it is certainly urgent to make available a baseline of pedigreed sequences for the species of Ostreidae. It would probably be desirable that the targeted barcode gene is the classical COI, but other candidates (e.g. the nuclear ITS2, see Salvi & Mariottini, Reference Salvi and Mariottini2012) may prove to be useful complements.
The Mediterranean specimens of small oysters collected at four localities in the Aegean Sea were morphologically so divergent that they could be a priori ascribed to different nominal species, all potential invaders in the semi-enclosed basin and, in fact, all recorded from the Mediterranean Sea by different authors. Actually, our specimens clearly belong to a single, morphologically highly variable species, included in an Indo-Pacific clade. This highlights the need for testing genetically the specimens used to record distinct species of small oysters as alien species in the Mediterranean (Çeviker, Reference Çeviker1999, Reference Çevik, Öztürk and Buzzurro2001; Sharon et al., Reference Sharon, Benayahu and Mienis2005; Zenetos et al., Reference Zenetos, Konstantinou and Konstantinou2009, Reference Zenetos, Katsanevakis, Poursanidis, Crocetta, Damalas, Apostolopoulos, Gravili, Vardala-Theodorou and Malaquias2011; Mienis et al., Reference Mienis, Rittner, Rilov and Almog2012a, Reference Mienis, Zaslow and Rittnerb; Crocetta et al., Reference Crocetta, Bitar, Zibrowius and Oliverio2013b). Unfortunately, with the sole exception of records published in Zenetos et al. (Reference Zenetos, Katsanevakis, Poursanidis, Crocetta, Damalas, Apostolopoulos, Gravili, Vardala-Theodorou and Malaquias2011: partim from Astypalaia, from a population hereby investigated), most of the specimens were recorded as empty shells, or their soft parts were not properly preserved, therefore making impossible a molecular check.
Marine alien species feature among the qualitative descriptors of good environmental status in the European Union's Marine Strategy Framework Directive. In the case of framework builder species, such as oysters (Krŭić, Reference Krŭić, Goffredo and Dubinsky2014), when they become invasive with high density in the invaded areas, their bearing on autochthonous communities may be remarkable. Therefore, alien species' inventories play important roles in informing regional policy and management decisions, as well as in identifying resource allocation priorities. The scientific community is called upon to pay particular attention to their accuracy and veracity (Ojaveer et al., Reference Ojaveer, Galil, Minchin, Olenin, Amorim, Canning-Clode, Chainho, Copp, Gollasch, Jelmert, Lehtiniemi, McKenzie, Mikuš, Miossec, Occhipinti-Ambrogi, Pećarević, Pederson, Quilez-Badia, Wijsman and Zenetos2014), and genetic data are often the only tool to make them reliable.
ACKNOWLEDGEMENTS
Paolo Crovato (Italy), Roland Houart (Belgium) and Henk Mienis (Israel) offered literature support; Maria Vittoria Modica (Italy) helped with laboratory work; we are grateful to all of them.
Supplementary materials and methods
The supplementary material for this article can be found at http://www.journals.cambridge.org/MBI
FINANCIAL SUPPORT
D.S. is supported by the post-doctoral fellowships SFRH/BPD/66592/2009 by the Fundação para a Ciência e Tecnologia (Portugal) and by the project ‘Genomics and Evolutionary Biology’ co-financed by North Portugal Regional Operational Program (ON.2) under NSRF through the European Regional Development Fund.