


INTRODUCTION
Dystrophinopathies are muscle diseases caused by the absence or abnormal expression of the protein dystrophin. Phenotypic presentation of dystrophinopathy has a degree of variability, but the clinical course is generally consistent across individuals. The complete absence of dystrophin results in the most severe form of the disease known as Duchene muscular dystrophy (DMD) and the milder form of the disease called Becker muscular dystrophy (BMD) is associated with partial but functional dystrophin production (Muntoni, Torelli, & Ferlini, Reference Muntoni, Torelli and Ferlini2003). Physical phenotype with rapid progression of muscle weakness resulting in eventual gait cessation is indicative of DMD, whereas prolonged ambulation is a distinguishing feature of the less severe BMD (Bushby et al., Reference Bushby, Finkel, Birnkrant, Case, Clemens, Cripe and Constantin2010).
In the brain, dystrophin is localized to both neurons and glia and it appears to play a role in both the development and function of brain structures (Lidov, Reference Lidov1996). Dystrophin is normally localized in the cerebral cortex, hippocampus, and cerebellum where it may serve to anchor molecules for neuronal function (Lidov, Reference Lidov1996), organize gamma-aminobutyric acid A and acetylcholine receptors (Cohen, Quarta, Fulgenzi, & Minciacchi, Reference Cohen, Quarta, Fulgenzi and Minciacchi2015), and stabilize postsynaptic areas. The development of the brain in the absence of dystrophin likely defines the pattern of cognitive and behavioral impairments that are observed in children with dystrophinopathy (Cohen et al., Reference Cohen, Quarta, Fulgenzi and Minciacchi2015; Lidov, Reference Lidov1996).
Neurocognitive impairments independent of motor declines have been identified within the dystrophinopathy population. There is an increased risk of overall intellectual disability; however, most children with dystrophinopathy have IQ within the normal range (Cotton, Voudouris, & Greenwood, Reference Cotton, Voudouris and Greenwood2005). Aspects of memory, visuospatial processing, and crystallized language abilities have all been documented to develop appropriately in children with dystrophinopathy (Hinton, De Vivo, Nereo, Goldstein, & Stern, Reference Hinton, De Vivo, Nereo, Goldstein and Stern2001; Hinton, Fee, Goldstein, & De Vivo, Reference Hinton, Fee, Goldstein and De Vivo2007). However, attention and executive functioning as well as linguistic processing are susceptible to impairments, although the findings are somewhat variable across studies. The most consistent finding across the literature is impaired digit span (Hinton et al., Reference Hinton, De Vivo, Nereo, Goldstein and Stern2001; Hinton, DeVivo, Fee, Goldstein, & Stern, Reference Hinton, DeVivo, Fee, Goldstein and Stern2004; Hinton et al., Reference Hinton, Fee, Goldstein and De Vivo2007; Leaffer, Fee, & Hinton, Reference Leaffer, Fee and Hinton2016; Wicksell, Kihlgren, Melin, & Eeg-Olofsson, Reference Wicksell, Kihlgren, Melin and Eeg-Olofsson2004; Wingeier et al., Reference Wingeier, Giger, Strozzi, Kreis, Joncourt, Conrad and Steinlin2011).
Evidence from parent behavior ratings indicate increased rates of attention, executive, and social deficits (Banihani et al., Reference Banihani, Smile, Yoon, Dupuis, Mosleh, Snider and McAdam2015; Donders & Taneja, Reference Donders and Taneja2009; Hendriksen & Vles, Reference Hendriksen and Vles2008; Hinton, Nereo, Fee, & Cyrulnik, Reference Hinton, Nereo, Fee and Cyrulnik2006) among children with dystrophinopathies. Hinton and colleagues have posited a core deficit in verbal working memory that may explain the cognitive profile (Cyrulnik et al., Reference Cyrulnik, Fee, Batchelder, Kiefel, Goldstein and Hinton2008; Hinton et al., Reference Hinton, DeVivo, Fee, Goldstein and Stern2004), whereas others have suggested it may reflect more generalized language or executive deficits (Donders & Taneja, Reference Donders and Taneja2009; Mento, Tarantino, & Bisiacchi, Reference Mento, Tarantino and Bisiacchi2011; Wicksell et al., Reference Wicksell, Kihlgren, Melin and Eeg-Olofsson2004).
Full-length dystrophin (Dp427) and smaller isoforms including Dp71 and Dp140 have been identified as having a role in cognitive function (Daoud et al., Reference Daoud, Angeard, Demerre, Martie, Benyaou, Leturcq and Tuffery2009; Taylor et al., Reference Taylor, Betts, Maroulis, Gilissen, Pedersen, Mowat and Buckley2010). Mutations typically distal of exon 63 and affecting the expression of the isoform Dp71 are associated with severe intellectual impairments (Daoud et al., Reference Daoud, Angeard, Demerre, Martie, Benyaou, Leturcq and Tuffery2009; Ricotti et al., Reference Ricotti, Mandy, Scoto, Pane, Deconinck, Messina and Muntoni2016). Furthermore, in addition to higher rates of intellectual disability, mutations disrupting Dp 71 have been associated with behavioral difficulties and impaired working memory (Ricotti et al., Reference Ricotti, Mandy, Scoto, Pane, Deconinck, Messina and Muntoni2016).
Mutations localized to exon 44–45 affecting Dp140 have also been associated with cognitive impairments, but the findings are less robust (Wingeier et al., Reference Wingeier, Giger, Strozzi, Kreis, Joncourt, Conrad and Steinlin2011). Thus, the data suggest that mutation position (distal of exon 63, exons 44–45) disrupts production of particular brain dystrophin isoforms (Dp71, Dp140), and this absence of isoform then, in turn, impacts brain function and cognitive performance. More detailed analysis of the association of mutation position with specific cognitive deficits is warranted.
Overall, the pattern of deficits observed among children with dystrophinopathy highlights specific areas of weakness that may impact the efficiency of cognitive processing and behavior, suggestive of deficiencies in executive functions. To further test this, we administered a battery of tests examining a range of executive skills (including measures of attention, set shifting, working memory, processing speed, and measures of executive deficits in everyday life) to a diverse cohort of 50 boys with dystrophinopathy. The goals of the study were to determine if individuals with dystrophinopathy have generalized executive difficulties in both cognitive processing and behavior that contribute to overall function, and to investigate if there is an association between this cognitive profile and mutation position affecting central nervous system isoforms.
Three hypotheses were tested. As a group, boys with dystrophinopathy will have lower performance on tasks of executive functions and higher parental ratings of problem executive behaviors than expected from the normative population. Furthermore, when each boy’s performance on executive measures is compared to his own general intellectual level, boys with dystrophinopathy will perform more poorly on executive than intellectual tasks. Lastly, when grouped by mutation position, boys with dystrophinopathy who have more distal mutations (disrupting Dp71 and/or Dp140 production) will have lower performance on all measures, including executive functions, than those with more proximal mutations.
METHODS
Sample
The dystrophinopathies are X linked disorders, thus males were recruited given the disorder is rare in females. Participants were recruited from 128 cases with dystrophinopathy routinely seen at the Pediatric Neuromuscular Center/MDA clinic at New York Presbyterian Hospital, associated with Columbia University. The clinic serves the greater New York metropolitan area and a wide range of socioeconomic levels. Inclusion criteria were: genetically confirmed diagnosis of dystrophinopathy, between 5 and 17 years of age, English as primary language of participant (not of parents), willingness to participate, and in relatively good health with no comorbid medical diagnosis apart from the diagnosis of dystrophinopathy. Seventy-two boys with dystrophinopathy met inclusion criteria (56 were out of the age ranges, 17 failed contact due to relocation or a wrong telephone number, and 5 refused participation). Fifty boys thus agreed to participate, 45 participants were diagnosed with Duchenne muscular dystrophy and 5 participants had the milder Becker muscular dystrophy.
Glucocorticoid therapy is the recommended treatment and 64% of participants were on this treatment, but no other reported medication. On a self-report developmental history completed by parents, seven participants (14%) were described with attention deficit hyperactivity disorder, two participants (4%) a spectrum disorder, and 16 participants (32%) were reported by parents as having delays in meeting developmental milestones. Income groups were calculated by annual family income and number of individuals in the home compared to the median family income in New York City. Groups were categorized by annual income as low for those making less than or equal to $35,000, middle income as those making between $35,000 and $99,999, and high as those making greater than or equal to $100,000.
Procedures
The study was approved by the institutional review board at Columbia University Medical Center (IRB #AAAA5627) and The Graduate Center of City University of New York and was supported by a grant from the Muscular Dystrophy Association. Members of the clinic team discussed potential participation with 55 individuals who met criteria and attended a clinic appointment. The study coordinator followed up and described the protocol in depth with interested families and 50 families agreed to participate. All parents or guardians gave informed consent and all participants gave assent before enrollment. Evaluations took place in a quiet room in the clinic, breaks were given as needed, and each assessment took approximately 2 hr.
All neuropsychological measures were administered to participants in a standardized order. Measures were chosen to assess a broad range of intellectual function and have minimal motor demands. All data were scored and converted to standardized scores. Data were coded without links to identifying information and stored in a secure database. The protocol adhered to the Health Insurance Portability and Accountability Act and IRB regulations to ensure patient confidentiality.
Assessment measures
Motor function
The Brooke and Vignos Scale was used to assess motor functioning (Brooke et al., Reference Brooke, Griggs, Mendell, Fenichel, Shumate and Pellegrino1981; Vignos, Spencer, & Archibald, Reference Vignos, Spencer and Archibald1963). Both measures have been shown to be reliable functional classification scales with high intraclass correlations and high interrater reliability for assessing individuals with muscular dystrophy (Lue, Lin, Chen, & Lu, Reference Lue, Lin, Chen and Lu2009; Mazzone et al., Reference Mazzone, Vasco, Palermo, Bianco, Galluccio, Ricotti and Mercuri2012). The scales include measurements of upper and lower extremity functioning. For this study, scores were combined to establish an overall motor functioning score and categorized into minimal impairment (1–5); able to walk, climb stairs, and full use of arms, moderate (6–12); able to walk, but unable to climb stairs and not able to raise hands over head; and severe impairment (13–16); wheelchair bound, limited use of upper extremities. Fine motor abilities were also assessed using a finger-tapping task to ensure participants were able to respond accurately using a computer keyboard.
The Finger Tapping Test has good test–retest reliability (.58 to .93) and good concurrent validity (.78 correlation with other measures that require precise finger movements), with minimal practice effects and good normative data for children based on several normative studies (Strauss, Sherman, & Spreen, Reference Strauss, Sherman and Spreen2006). Using a standardized counting device, participants were instructed to tap their index as quickly as possible in 10 s. Each motor measure was administered by, or supervised by, a licensed physical therapist.
Intellectual function
Participants completed two measures that were used as proxies for general intellectual function: The Peabody Picture Vocabulary Test-IV (PPVT-IV) (Dunn & Dunn, Reference Dunn and Dunn2007), a measure of single word comprehension, and the Comprehensive Test of Nonverbal Intelligence-2 (CTONI-2), a nonverbal measure of analogical reasoning, categorical classification, and sequential reasoning. The tests were chosen to offer an estimation of crystallized intellectual function within the verbal and visuospatial domains, while removing the contributions of motor and working memory confounds associated with many standardized tests of IQ, as well allowing for a wider age range.
Because the children have known motor involvement, subtests relying of speed of written responses were deemed inappropriate. And because one of the primary outcome measures was working memory, intellectual function was estimated using tests that did not rely on those skills. Both the PPVT-IV (Pearson correlation coefficient of .71) and the CTONI-2 (criterion validity, Pearson correlation of .81) are strongly correlated with the Full Scale IQ score of the Wechsler Intelligence Scales for Children (Craig & Olsen, Reference Craig and Olson1991; Rossen, Shearer, Penfield, & Kranzler, Reference Rossen, Shearer, Penfield and Kranzler2005). Scores on the PPVT-4 and CTONI-2 were combined and the mean was used to create a composite score of crystallized intellectual function (IF).
Attention/executive measures
Primary measures were chosen from The National Institutes of Health Toolbox (Weintraub et al., Reference Weintraub, Bauer, Zelazo, Wallner-Allen, Dikmen, Heaton and Gershon2013). The entire battery is computerized with automated scoring and generates age-adjusted standardized scores. Normative data for this set of measures is based on a large national sample of 4859 participants between the ages of 3 and 85 that are representative of the U.S. population based on gender, race/ethnicity, and socioeconomic status. Studies have been conducted across various age ranges, including children between 3 and 15 years of age (Zelazo & Bauer, Reference Zelazo and Bauer2013), and the measures have been validated when compared to well-established neuropsychological measures.
The Toolbox tasks are standardized cognitive instruments that have been used on pediatric populations (Casaletto et al., Reference Casaletto, Umlauf, Beaumont, Gershon, Slotkin, Akshoomoff and Heaton2015; Weintraub et al., Reference Weintraub, Bauer, Zelazo, Wallner-Allen, Dikmen, Heaton and Gershon2013). Selected subtests from the NIH Cognitive Toolbox were administered, including those indexing attention (Flanker Inhibitory Control Test), set shifting (Dimensional Change Card Sort Test), working memory (List Sorting Working Memory Test), and processing speed (Pattern Comparison Processing Speed Test).
The Flanker task measures selective attention/inhibitory control and requires the participant to focus on a stimulus while inhibiting congruent and incongruent stimuli. Accuracy and reaction time are measured. The Dimensional Change Card Sort task measures mental flexibility. Two dimensions are assessed, including both the color and shape of an object, and require the ability to shift dimensional sets. Scoring is based on both accuracy and reaction time. The List Sorting Working Memory Test measures immediate recall and sequencing of different visually and orally presented stimuli (animals and foods) in size order from smallest to largest, first within a single dimension and then on two dimensions. The score is equal to the number of items recalled and sequenced correctly. The Pattern Comparison Processing Speed Test measures speed of processing. Participants are given 85 s to rapidly judge as many item sets as possible to determine whether two pictures presented are the same or not. Participants’ raw score is the number of correct items within 85 s.
The Digit Span subtest from the Wechsler Intelligence Scale for Children-IV (Wechsler, Reference Wechsler2004), was administered as a measure of verbal span and working memory. Participants were asked to repeat numbers in the same order, or in the reverse order, of presentation. Total score reflects performance on the entire measure, and individual maximum span length of both forward and backward administration was also calculated.
All raw scores were age standardized based on test normative data from general population means and standard deviations, and then converted to Z-scores.
Behavioral assessment
Parent ratings of children’s executive behaviors and self-regulation in everyday life were assessed using the Behavior Rating Inventory of Executive Function (BRIEF) (Gioia, Isquith, Guy, & Kenworthy, Reference Gioia, Isquith, Guy and Kenworthy2000). The scale is designed for children 5 to 18 years of age and has been shown to have clinical utility, as well as predictive validity, for examining executive behaviors in everyday life. The test is reported to have strong validity, test–retest reliability (.79 to .88), and internal consistency (Cronbach’s alpha ranging from .80 to .98). There is an overall Global Executive composite score, as well as two broad scales of Metacognition and Behavioral Regulation, and eight theoretically derived scales that measure different aspects of executive functioning (Inhibit, Shift, Emotional Control, Initiate, Working Memory, Plan/Organize, Organization of Materials, and Monitor).
The scale was administered in either English or Spanish, depending on parent’s or guardian’s stated primary language. Raw scores were age standardized to T scores, and higher T scores indicate greater executive difficulties. Ratings were dichotomized based on a T score cut-point of 65 and above (at or above the 94th percentile and indicative of clinically significant executive difficulties, as designated by the BRIEF manual) (Gioia et al., Reference Gioia, Isquith, Guy and Kenworthy2000).
Mutation position
Participant medical records were reviewed for the results of genome sequencing and dystrophin gene mutation analysis. Mutation positions were categorized into three groups: upstream of exon 30, exons 31–62 (disrupting Dp 140 production) and downstream of exon 63 (disrupting Dp 71 production).
Statistical Methods
Descriptive demographic analyses were done on standardized scores. Shapiro-Wilk analyses were run to ensure the data were normally distributed for parametric analyses, Box’s Test of Equality of Covariance assessed for homogeneity of variances, and a linear relationship was determined between the variables including the covariate. To further examine for potential motor confounds, the Brooke and Vignos Motor categorized groups’ (minimal, moderate, and severe) and performance on the Finger Tapping test (dichotomized by performance 1 SD below the population mean vs. above) was compared to performance on all cognitive measures. Bonferroni alpha level was used to adjust for the number of comparisons to reduce the probability of Type I error.
Part 1. Cognitive battery: attention/executive functioning
t Tests were used to examine cognitive performance between the group of boys with dystrophinopathy and expected normative values. To examine whether performance on tests of attention, processing speed, working memory, and executive functioning was significantly different from general intellectual level, paired t tests were conducted to compare performance on each executive task with individual IF composite estimates. Bonferroni adjusted alpha was set at .007 (.05/7 comparisons) to reduce the probability of Type I error.
Behavioral Assessment
To determine whether parent reported rates of executive problems were greater than those expected within the general population, the frequency of participants scoring in the “clinically significant” range (T > 65) on each of the scales was determined and compared to the expected frequencies on the measure (based on the normative sample) using chi-square statistics. The stringent adjusted alpha at .007 was used to reduce the probability of Type I error.
Part 2. Mutation position and cognition
To investigate the association between mutation position and cognition, performance on all neuropsychological measures was compared between the three groups. A multivariate analysis of covariance (MANCOVA) was conducted to determine differences in cognitive performance between the three groups while controlling for the potential effect of motor functioning. Bonferroni procedure post hoc analyses were conducted to examine specific significant differences between groups on the cognitive measures. Chi-square statistics were used to compare significantly elevated BRIEF scales across the groups. Bonferroni adjusted alpha level was set at .006 to reduce the probability of Type I error.
RESULTS
Demographic information is presented in Table 1. Participants came from diverse backgrounds with a broad range of socioeconomic status and education. There were no significant differences between the groups across demographic variables. It is noteworthy, however, that there is a trend to suggest a greater percentage of those with low income have downstream mutations.
Table 1 Descriptive characteristics of the total sample

MFI=median family income.
The motor scale identified 38% of the sample as severely impaired, the majority of participants were minimally impaired (56%), but when performance on neuropsychological test measures was compared across the three motor categories (minimal, moderate, and severe), there were no significant differences (F range =.01 to 2.80; p > .01). Similarly, although 29% of the sample scored below 1 SD from the population mean on the Finger Tapping test, there was no difference on all cognitive measures when the groups were compared (F range =.01 to 4.23; p > .01). These results suggest that participants’ performance on the cognitive tests was unlikely confounded by motor limitations.
For subsequent analyses, all assumptions were met for parametric analysis; cognitive data were normally distributed with equality of variances.
Part 1. Cognitive Battery: Attention/Executive Functioning
Neuropsychological performance data are presented in Table 2.
Table 2 Neuropsychological mean performance standardized data

* p < .007.
PPVT=Peabody Picture Vocabulary Test-IV; CTONI=Comprehensive Test of Nonverbal Intelligence-2, IF=intellectual functioning.
Normative comparisons
Mean performance on measures of single word comprehension and perceptual reasoning was comparable (t(50) = −.94; p = .35). Probands’ sores on each measure fell in the average range, and neither was significantly different from expected normative values. IF composite estimates ranged from delayed to high average and mean performance was in in the average range (z = −.34; equivalent to a standard score of 95). Probands’ mean performance on all attention/executive function measures was significantly lower than the normative values (see Table 2).
Selected tasks of attention/executive function
Comparison of standardized scores on Toolbox tasks and on the Digit Span subtest revealed no significant differences between measures.
In contrast, all executive tasks were significantly lower than estimated IF, with the biggest differences observed on the Flanker, List Sorting Working Memory Task and Digit Span total subtests (see Table 2; Figure 1). When Digit Span was broken down into maximum span forward and maximum span backward, maximum forward span did not differ from estimated IF (effect size Cohen’s d = .37).
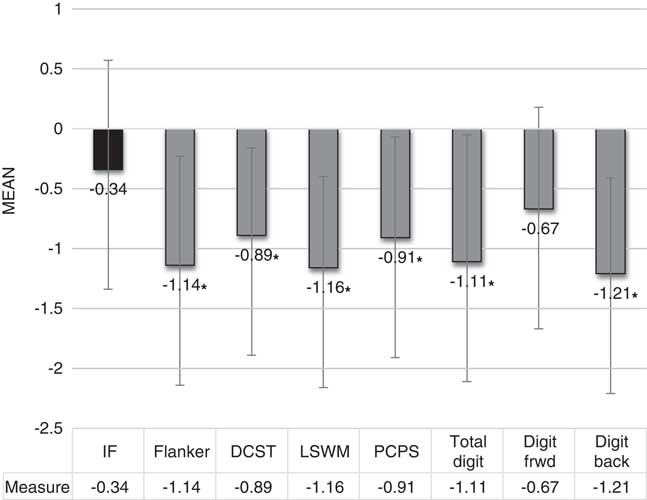
Fig. 1 Mean performance on all executive measures was significantly lower than the IF estimate. * = p < .007. Error bars = standard deviation. IF = intellectual functioning; DCST = Dimensional Change Card Sort Test; LSWM = List Sorting Working Memory Test; PCPS = Pattern Comparison Processing Speed Test; Total digit = Total Digit Span test; Digit frwd = Digit Span forward test; Digit back = Digit Span backward test.
Executive behavior
Only 44 parents completed the BRIEF scales. Thirty-five completed it in English and 9 completed it in Spanish, based on the parent or guardian’s primary language. When age, IF, motor abilities, and all executive measures from the six families whose scales were not completed were compared to those with completed scales, no differences were observed, suggesting the missing data are reflective of the overall sample.
The percentages of participants who scored above clinical cutoff of T > 65 for each scale on the BRIEF are presented in Table 3. Parents were more likely to rate their son with dystrophinopathy as having clinically significant difficulties with executive control on the Shift, Emotional Control, and Behavior Regulation indices than expected.
Table 3 BRIEF executive scale T scores in a sample of 44 with dystrophinopathy

a Higher frequency than expected, >6% of population.
* p < .007.
BRIEF = Behavior Rating Inventory of Executive Function.
Part 2. Mutation Position and Cognition
Comparison between the three mutation position groups while controlling for the impact of motor abilities revealed a significant difference between the groups (F(8,34) = 26.38; p = .000; Wilks’ Λ = .139). IF and Total Digit Span were the only two variables found to be significantly different between the groups. Bonferroni post hoc results revealed those with mutations downstream of exon 63 (n = 7) had significantly poorer mean performances on the IF estimate (p = .005) and Total Digit Span (p = .003) than those with a mutation positioned upstream of exon 30 (Figure 2). Notably, Digit Span forward also approached significance (p = .028). However, there were no other significant differences including performances on IF and Digit Span between those with mutations downstream of exon 63 and those with mutations in the exons 31–62 group (Table 4). There were also no significant differences between the mutation position groups on elevated BRIEF Shift, Emotional Control and Behavior Regulation scales.
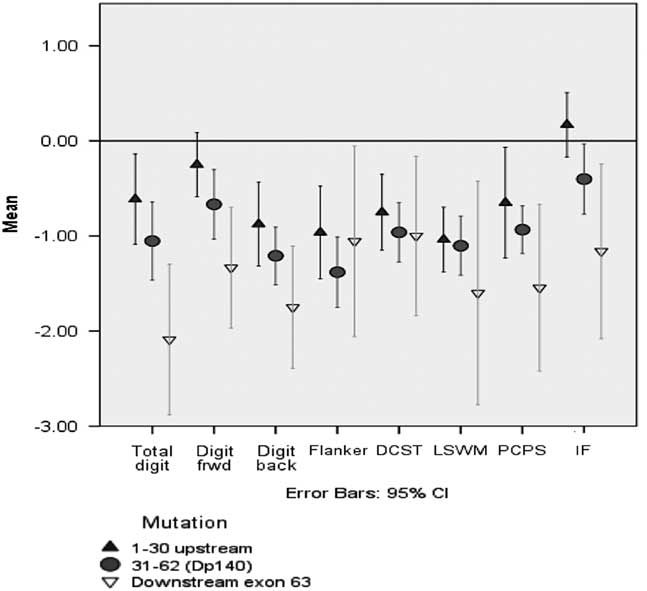
Fig. 2 Comparison of cognitive performance between the mutation position groups.
Table 4 Between mutation position group comparisons with motor functioning as a covariate

* p < .006.
IF = intellectual functioning; BRIEF = Behavior Rating Inventory of Executive Function; WNL = within normal limits.
DISCUSSION
Our study set out to examine a diverse sample of individuals with dystrophinopathy to determine whether deficits in executive functions underlie their unique neuropsychological profile and to examine the association between these functions and mutation position. We tested three hypotheses. First, we tested whether the group performed more poorly than normative samples on a range of measures of executive ability and parental ratings of problem executive behaviors. Next, we examined whether within the sample of boys with dystrophinopathy, participants performed more poorly on executive measures than they did on intellectual function. Lastly, we examined whether mutation position contributed to performance by grouping participants according to mutation location and testing whether those in the group with more distal mutations (disrupting Dp71 and/or Dp140 production) had lower performance on cognitive measures, than those in groups with more proximal mutations.
Results indicated that on direct testing, performance across all aspects of executive function studied was both lower than expected when compared to standardization samples and lower than expected when compared to participants’ intellectual functioning. Lower performance was found across measures assessing attention, executive control, set shifting, processing speed, and working memory, when compared to crystallized abilities reflective of intellectual level. Executive deficits also manifested in the performance of everyday functional activities. Parents from the sample rated their children as having limited flexibility, difficulty transitioning from one activity to the next, and poor emotion regulation. Thus, among children with dystrophinopathy, there is a generalized deficit in executive functioning.
Our study also explored the association between genotype and the neuropsychological profile. It should be noted that these analyses are limited by a small number of participants (n = 7) with mutations downstream of exon 63; thus, our negative findings need to be interpreted with caution as we may not have had adequate power to detect smaller effects. Nonetheless, our positive results were consistent with prior findings, adding credence to the results. Overall IF and performance on Digit Span were both significantly lower for those individuals with distal mutations. Our results replicate the findings of Ricotti et al. (Reference Ricotti, Mandy, Scoto, Pane, Deconinck, Messina and Muntoni2016) that performance in the Working memory index that includes Digit Span was significantly lower for those with mutations downstream of exon 63 affecting Dp 71.
We found performance on Digit Span was significantly lower for those individuals with distal mutations, suggesting a possible genotypic association with a specific cognitive deficit. We did not, however, find any association of performance on those mutations that disrupt Dp140, contrary to other reports (Wingeier et al., Reference Wingeier, Giger, Strozzi, Kreis, Joncourt, Conrad and Steinlin2011). However, it is noteworthy that across all the cognitive measures there is a consistent trend in scores across the mutation groups such that mean scores from those with deletions between exons 31–62 (impacting Dp 140) fall in between those of the other two groups. The findings are not statistically significant, but the trend is intriguing. Moreover, the trend appears to be most pronounced on the Total Digit Span Z-score, where there is approximately a doubling of value across each mutation position. A much larger sample is necessary to rule out convincingly the possibility of subtle associations between cognition of mutations affecting Dp140.
Other research has inferred that Digit Span is not just a measure of basic attention, but an important predictor of verbal memory functioning (Hurlstone, Hitch, & Baddeley, Reference Hurlstone, Hitch and Baddeley2014). Furthermore, empirical evidence assessing the general population have found a strong correlation between Digit Span and general intelligence (Gignac & Weiss, Reference Gignac and Weiss2015). Greater span has been demonstrated to strongly influence greater cognitive abilities as well as in the application of knowledge in everyday functioning such as academics. The current findings suggest that performance on Digit Span is significantly lower for individuals with distal mutations. The data offer a tantalizing possibility that reduced span capacity may somehow be distinct from other executive tasks within this population and may contribute to decreased performance in other areas of cognition. Future studies with larger sample sizes are necessary to address this definitively.
Strengths of our study include the diversity of the sample; our cohort’s varied ethnicity and socioeconomic status is distinct from many samples described in the dystrophinopathy cognitive research. As such, the diversity in our sample is more representative of the population of those affected by the disorder. Additionally, the measures chosen for the study represent a greater variety of attention/executive skills (including selective attention, set shifting and cognitive flexibility, working memory, and processing speed) than have previously been tested in one study. Our simultaneous collection and analysis of motor data considering the influence of motor abilities on performance offer the best possible evidence that the cognitive data were likely not negatively influenced by motor functioning. And our choice of non-traditional measures of crystallized intellectual function within the verbal and visuospatial domains ensured there were no potential motor and working memory confounds.
Although the lack of comparison sample might be considered a weakness of the study, the data were investigated using each participant as his own control, thus strengthening the validity of our findings. By using paired t tests, the standardized executive performance for each participant was compared directly to his own standardized IF performance. Finally, sample size was relatively large for the studied rare population and adequate for the aims of the study to detect clinical significance for the primary outcomes. However, when the sample was broken into smaller groups for the mutational analyses, data were limited, thus decreasing the power to detect more subtle significant differences. As a result, the negative findings should be considered mostly exploratory.
For the behavior rating comparison, the sample was compared to published norms. It is possible that the general effects of chronic illness, not specific to dystrophinopathy, might impact on parent report of executive skills in everyday functioning, yet it is known that, among other groups with chronic illness (such as diabetes), executive functions are not always reported to be elevated (Duke & Harris, Reference Duke and Harris2014), so we can speculate that our data reflect an increased level of difficulty with executive behaviors in boys with dystrophinopathy.
In conclusion, our findings demonstrate children with dystrophinopathy are at increased risk for having generalized executive deficits. Since well-developed executive skills have been linked to protective health behaviors, reduced risk behaviors, and greater longevity (especially in those with chronic illness), ensuring children receive targeted interventions to help improve their executive control will impact positively on day-to-day functioning and improve overall quality of life.
ACKNOWLEDGMENTS
We are very grateful to the families who took the time and effort to participate in our project. Special thanks to Sally Dunaway, PT, DPT, Ashwini K. Rao, OTR, EdD, Mercedes Vega Villar, Fiona McMahon, Ruta Patel, and Justine Payne for their contribution to the physical therapy assessments. This work was supported by a grant from the Muscular Dystrophy Association to V.J.H. Disclosures: Fee, R.J., Montes, J, and Hinton, V.J. declare that they have no conflicts of interest.






