


Introduction
Dyskeratosis congenita is a rare genetic disorder which presents with the classical triad of skin pigmentation, nail dystrophy and mucosal leukoplakia in 80–90 per cent of patients.Reference Cole, Rauschkolb and Toomey1, Reference Kirwan and Dokal2 The condition was first described in 1906 by Zinsser and later by Engman and Cole, and is thus also known as Zinsser–Cole–Engman syndrome.Reference Cole, Rauschkolb and Toomey1, Reference Zinsser3, Reference Engman4
Gene studies have shown that dyskeratosis congenita belongs to a group of diseases characterised by impaired telomerase function. This predisposes individuals with this condition to premature mortality due to malignant transformation, bone marrow failure or pulmonary disease.Reference Garcia, Wright and Shay5
Case report
A 30-year-old man was referred to our ENT department with a two-month history of a lesion on the posterior pharyngeal wall. This was asymptomatic and had been detected incidentally by the patient.
Oral examination revealed an ulcerated, granular lesion visible on the posterior pharyngeal wall and extending horizontally across the full width of the posterior pharynx.
The lesion was biopsied under general anaesthesia. Histological analysis identified it as a moderately differentiated squamous cell carcinoma.
Cross-sectional imaging established a tumour (T) node (N) staging of T2 N0.
As a child, the patient had originally been under the care of the dermatologists for ectodermal dysplasia when the diagnosis of dyskeratosis congenita had been made (Figure 1). Subsequent visits over the years had included presentations with various oral lesions, biopsies of which had shown chronic inflammation. Several years later, when the patient was in his twenties, oral ulceration developed which was also histologically benign and treated with topical steroids.
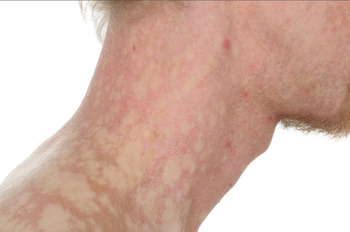
Fig. 1 Clinical photograph of the presented patient, showing the characteristic skin appearance of dyskeratosis congenita. © Oxford medical illustration (OMI).
The current presentation was discussed by the multi-disciplinary team, and a consensus decision made to undertake surgical treatment in the form of a transoral laser resection. This was undertaken uneventfully, and the patient recovered well.
However, post-operative histological analysis of the surgical specimen confirmed involved resection margins. Following further multi-disciplinary team discussion, further surgical resection was recommended. This was performed successfully, with clear margins.
At the time of writing, the patient was disease-free and remained under regular follow up.
Discussion
Dyskeratosis congenita is a rare, progressive, inherited, multisystem disorder which may be X-linked, autosomal dominant or recessive.Reference Garcia, Wright and Shay5, Reference Handley, McCaul and Ogden6 The majority of sufferers are male, suggesting that X-linked patterns of inheritance are most common.Reference Dokal7 The prevalence is less than one per million. Patients typically appear healthy at birth but develop mucocutaneous features later in life. A wide range of somatic problems have also been described, including bone marrow, pulmonary, gastrointestinal, endocrine, skeletal, urological, immunological and neurological abnormalities.Reference Handley, McCaul and Ogden6 Bone marrow failure is the leading cause of death (60–70 per cent), followed by pulmonary disease (10–15 per cent) and malignancy (10 per cent), with death occurring at a median age of 16 years.Reference Dokal7, Reference Walne and Dokal9
Telomere dysfunction plays a principal role in the pathogenesis of dyskeratosis congenita.Reference Garcia, Wright and Shay5 Under normal physiological conditions, telomerase plays fundamental roles in maintaining the replicative ability of progenitor cells in telomerase-expressing tissues that undergo renewal. There has been much interest in dyskeratosis congenita because ‘telomere maintenance’ is closely associated with important life events, including ageing and cancer predisposition.Reference Nishio and Kojima10 A defect in telomerase function can result in cell-cycle arrest and consequently cell senescence in normally proliferative tissues. Furthermore, cells with impaired telomerase function demonstrate significant genetic instability and can undergo malignant transformation.Reference Marrone, Walne and Dokal11 The most frequent solid tumours are head and neck squamous cell carcinomas, followed by skin and anorectal cancers.Reference Alter, Giri, Savage and Rosenberg12 It has been reported that dyskeratosis congenita patients have an 11-fold higher cancer risk compared with the general population.Reference Alter, Giri, Savage and Rosenberg12
Oral features are common and include: hypodontia; delayed dental eruption; short, blunt tooth roots; extensive dental caries; gingival inflammation and bleeding; and loss of alveolar bone and buccal mucosa with leukoplakia and irregular ulcers.Reference Wald and Diner13, Reference Yavuzyilmaz, Yamalik, Yetgin and Kansu14 Patients with dyskeratosis congenita have an increased risk of developing squamous cell cancers due to malignant transformation of oral lesions; such tumours often arise from pre-existing leukoplakia, with a peak incidence (approximately 35 per cent) in the third decade.Reference Cannell15, Reference Sirinavin and Trowbridge16 A proposed sequence of events for the development of carcinomas from such lesions has been suggested (see Table I).Reference Cannell15
Table I Proposed carcinogenesis of oral tumours

Adapted from cannell (1971).Reference Cannell15 Y = years
Histologically, squamous cell carcinoma is the most common malignancy arising in dyskeratosis congenita.Reference Davidson and Connor17 Over the past 20 years, there have been four published cases of head and neck carcinoma arising in patients with dyskeratosis congenita, detailed in Table II.
Table II Head and neck SCC arising in dyskeratosis congenita: published cases

SCC = squamous cell carcinoma; RT = radiotherapy
Malignancy in these patients is generally considered to carry a poor prognosis. Diagnosis is often delayed as the tumour tends to originate in an area of persistent leukoplakia; there are also high rates of local recurrence and multiple malignancy.Reference Handley, McCaul and Ogden6, Reference Baykal, Kavak, Gülcan and Büyükbabani22
The management of such malignancies can also be challenging. This is particularly relevant in the case of head and neck cancers. In general cases of such cancers, there is an increasing trend for the use of primary chemoradiotherapy regimens, with surgery reserved for salvage, to facilitate organ preservation. However, in patients with dyskeratosis congenita the application of conventional DNA-damaging therapies such as irradiation and/or chemotherapy could potentially accelerate the processes of bone marrow failure, pulmonary disease and/or carcinogenesis.Reference Handley, McCaul and Ogden6, Reference Komune, Hara, Tamae, Izu, Tokura and Joe18 There is also the risk of increased morbidity in dyskeratosis congenita patients, as the impaired regeneration of mucosal linings may predispose these patients to severe, prolonged mucositis following radiation therapy.Reference Cengiz, Celebioglu, Ozyar and Atahan19 There is also the risk that the immunosuppression associated with DNA-damaging therapies will aid the systemic spread of existing cancer cells and the development of multiple cancers, a feature often observed in dyskeratosis congenita.Reference Handley, McCaul and Ogden6, Reference Komune, Hara, Tamae, Izu, Tokura and Joe18 It was for these reasons that our patient's multi-disciplinary team considered surgical treatment to be the primary modality.
• Dyskeratosis congenita is a rare genetic disorder caused by impaired telomerase function
• Presentation classically involves skin pigmentation, nail dystrophy and mucosal leukoplakia
• Patients die prematurely due to malignancy, bone marrow failure or pulmonary disease
• Malignancy has a poor prognosis
• Management is largely surgical; DNA-damaging therapies should be avoided
At present, there is no curative treatment for dyskeratosis congenita. However, early diagnosis may allow preventative measures to be taken. Regular observation and biopsy of suspicious oral lesions is recommended to detect malignant change. Topical steroids may also be useful for leukoplastic lesions that are inappropriate for excision. Telomerase inhibitors represent a potential form of treatment, and there are some promising compounds under investigation, but inefficient delivery appears to be a limitation. There is some hope that nanomedicine may help to solve this delivery problem; if not, alternative delivery technologies may be required to overcome this complex and fatal disease.
Conclusion
Although dyskeratosis congenita is a rare condition, early identification is vital given its fatal consequences and risk of malignant progression. There is a risk of head and neck squamous cell carcinoma developing within pre-existing oral lesions. The presented case highlights the importance of avoiding DNA-damaging therapies such as radiotherapy, and illustrates the use of surgery as primary treatment for such patients.
Acknowledgement
We would like to thank Dr C Alcock FRCR for providing an invaluable oncological perspective on this paper.



