


Introduction
Congenital external auditory canal atresia has been classified by Schuknecht into four types.Reference De la Cruz, Fayad, Nadol and McKenna1
Congenital canal stenosis is a milder form of canal atresia within this classification system. It has been defined by Cole and Jahrsdoerfer as an external auditory canal with a diameter of 4 mm or less.Reference Cole and Jahrsdoerfer2 These authors reported a series of 54 patients with congenital aural stenosis, almost half of whom had canal cholesteatoma.
Congenital canal atresia is relatively uncommon, with a prevalence of one to five in 20 000 live births. It can be associated with anomalies of the pinna and middle ear. This condition results from failure of the ectodermal cord to canalise. It is usually unilateral but can be bilateral. Essentially, there is an absence of bony meatus, and a plate of bone exists in place of the tympanic membrane.Reference Jafek, Nager, Strife and Gayler3 In cases of canal atresia, the presence of cholesteatoma may be indicated by the computed tomography scan.Reference Jahrsdoefer4
Materials and methods
We performed a retrospective review of all patients with congenital external auditory canal atresia and stenosis, together with cholesteatoma, treated at Universiti Kebangsaan Malaysia Medical Centre between 1998 and 2009.
Data collected included demographic data, presenting symptoms and clinical findings. The results of imaging studies and hearing assessments were included and analysed. Patients' surgical management and outcome data were also evaluated.
Results
We identified 41 cases of external auditory canal stenosis or canal atresia operated upon at Universiti Kebangsaan Malaysia Medical Centre. Seventeen of these cases (43.9 per cent) were associated with cholesteatoma. However, the medical records of four cases were unretrievable. The remaining 13 patients comprised five females (38.5 per cent) and eight males (61.5 per cent), with ages ranging from four to 73 years (mean age, 21 years).
Ten out of these 13 patients had a history of recurrent otorrhoea as their chief presenting symptom (Table I). Another two patients complained mainly of postauricular discharge from recurrent mastoid abscess. One patient had symptoms of otalgia, postauricular tenderness, headache, neck stiffness and reduced level of consciousness.
Table I Patients' presenting symptoms, external auditory canal State and cholesteatoma site

Pt = patient; y = years; EAC = external auditory canal; med = medial
Clinical findings revealed that six (46.2 per cent) of these patients had canal stenosis whilst the other seven had canal atresia. Of the seven patients with canal atresia, three had a history of mastoid abscess. Otorrhoea is generally the main symptom in canal stenosis cases; only one of our patients presented with mastoiditis associated with meningitis. Microtia was present as an associated anomaly in eight (61.5 per cent) patients (who mostly had canal atresia rather than stenosis); the remaining patients had a normal pinna. There was no documented facial nerve palsy in any of the patients.
Four patients had a history of mastoid abscess, and presented with a discharging wound in the postauricular area.
Another patient presented with otalgia associated with postauricular tenderness, headache, neck stiffness and reduced level of consciousness. He denied any history of otorrhoea. In this patient, otoscopic examination revealed a stenotic external auditory canal, with a poor view of the tympanic membrane. This patient was admitted to the medical ward and treated for meningitis. The diagnosis of meningitis was suspected on analysis of cerebrospinal fluid collected via lumbar puncture; the fluid was turbid, with the presence of lymphocytes and neutrophils. However, there was no growth of any pathogenic organism on microbial culture. Imaging studies revealed opacity of the left mastoid air cells, consistent with a diagnosis of acute mastoiditis. A small intracranial abscess was also seen. This patient received initial treatment with intravenous ceftriaxone, and later underwent left modified radical mastoidectomy.
Hearing was assessed by pure tone audiometry in all our patients (Table II). Ten patients (76.9 per cent) had conductive hearing loss, with air–bone gaps ranging from 20 to 80 dB (mean 39.8 dB). The other three patients had severe to profound sensorineural hearing loss.
Table II Summary of patient management and outcome

*Viewed intra-operatively. Pt = patient; y = years; HRCT = high resolution computed tomography; ABG = air–bone gap; pre-op = pre-operative; post-op = post-operative; F = female; M = male; SNHL = sensorineural hearing loss
High resolution computed tomography (CT) scanning of the temporal bone was also performed in all cases. Cholesteatoma was suspected on visualisation of a soft tissue mass with or without bony erosion; cholesteatoma was subsequently confirmed intra-operatively (Figures 1 and 2). The extent of the cholesteatoma sac determined the type of surgery required: small and limited cholesteatomas were treated via an endaural canalplasty approach, while extensive or complicated disease was treated with canal wall down mastoidectomy.

Fig. 1 Axial high resolution computed tomography scan showing right canal atresia, with the mastoid cavity filled with soft tissue density (single arrow) and bony erosion suggestive of cholesteatoma. There is also soft tissue swelling in the postauricular region (double arrows).
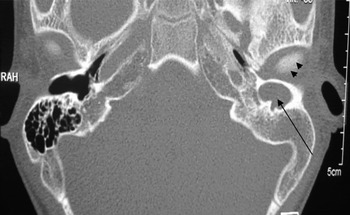
Fig. 2 Axial high resolution computed tomography scan of a patient with left canal atresia and a cholesteatoma sac medial to the atretic segment within the external auditory canal (arrow). Arrowheads indicate the temporomandibular joint. RAH = Temporomandibular joint
All patients with canal atresia or stenosis plus suspected cholesteatoma were managed surgically in our centre. Documented intra-operative findings indicated that the cholesteatoma sac was found medial to the atretic or stenosed external auditory canal, and extended into the middle ear or mastoid cavity. There was evidence of bony ear canal wall or ossicular erosion in eight cases. Five cases had ossicular abnormality, such as fused malleoincudal complex. In addition, dehiscence of the facial nerve was observed in three cases.
Six patients underwent canalplasty with split skin grafting. The other seven patients underwent modified radical mastoidectomy.
Six patients recovered well, without needing further surgery. Two (15.4 per cent) required repeated canalplasty because of soft tissue restenosis. The remaining five patients had granulation tissue within the mastoid cavity, which was successfully managed by cauterisation or split skin grafting in the clinic. There was no recurrence of cholesteatoma. The follow-up duration ranged from 10 months to 11 years.
The post-operative hearing outcome was documented in 12 patients. Three of the patients who had presented with severe to profound sensorineural hearing loss had similar hearing levels post-operatively. Of those with pre-operative conductive hearing loss, five patients achieved a good hearing outcome (i.e. within 30 dB).
Discussion
Congenital external auditory canal atresia is often associated with malformations of the middle and inner ear. Bony atresia is more common than membranous atresia.Reference Jahrsdoefer4 It is a rare condition, and 10 per cent of cases are associated with recognisable syndromes.Reference Ghosh, Saha, Sandhu and Saha5
The varying degrees of canal atresia result from arrested development of the external ear canal and middle ear in utero.Reference Caughney, Jahrsdoefer and Kesser6 Classifications of canal atresia by various authors include canal stenosis as a milder form of external auditory canal maldevelopment.Reference Glasscock, Schwaber, Nissen and Jackson7–Reference Altmann9
Cholesteatoma has been reported in cases of congenital canal anomalies.Reference Vrabec and Chaljub10 The risk of cholesteatoma is greater if there is a segment of the external ear canal with skin lining present medial to the atretic or stenosed canal.
In patients with congenital canal atresia, an underlying cholesteatoma must always be suspected. The likelihood of cholesteatoma is greater when patients present with recurrent infection, otorrhoea or acute facial palsy.Reference De la Cruz, Linthicum and Luxford8
Several theories have been proposed to explain the existence of congenital cholesteatoma, including the invagination theory, metaplasia theory and epidermoid formation theory. However, these theories apply only to cases of middle-ear cholesteatoma.Reference Michaels11
The embryonic cellular development arrest that results in canal atresia may suggest another mechanism for congenital cholesteatoma development. Cholesteatoma has been described lateral to the atretic plate on several occasions. Conceptually, cholesteatoma occurring lateral to the atretic plate is consistent with an arrest of external auditory canal development, leaving a small pouch or stenosed canal covered with ectoderm. This small pouch or stenosed ectoderm may grow into a cholesteatoma. The trapping of epithelium lateral to the atretic plate during embryological arrest of external auditory canal formation is probably the cause of cholesteatoma in cases of congenital canal atresia.Reference Caughney, Jahrsdoefer and Kesser6, Reference Sade, Babiacki and Pinkus12
When managing patients with congenital canal atresia, high resolution CT is routinely performed at the age of four to five years. At this age, temporal bone growth is adequate and mineralisation of the ossicles allows better visualisation.Reference Tasar, Yetiser, Yildirim, Bozlar, Tasar and Saglam13 Imaging studies also allow complete assessment of the middle-ear cavity, mastoid pneumatisation and inner ear status (the latter as regards candidacy for canalplasty). Mastoid pneumatisation and the presence of the stapes have been cited as the most important criteria for successful canal atresia surgery.Reference Ghosh, Saha, Sandhu and Saha5 Radiological studies also allow detection of cholesteatoma, indicating an urgent need for surgery.
High resolution CT scanning, both with and without contrast, is a reliable way of detecting cholesteatoma in cases of canal atresia. The keratin content of cholesteatoma gives it a lower density than brain tissue. In cases of congenital canal atresia with cholesteatoma, high resolution CT also plays a vital role in assessing the extent of the cholesteatoma, enabling better surgical planning, and in grading the likely post-operative hearing outcome. In our 11-year series of patients with congenital canal atresia, 17 of 41 patients (43.9 per cent) had associated cholesteatoma; this is similar to other authors' findings.Reference Cole and Jahrsdoerfer2
The role of magnetic resonance imaging (MRI) in detecting cholesteatoma has also been studied. Cholesteatomas have a low signal intensity on T1-weighted MRI scans and a high intensity on T2-weighted scans, and do not enhance with gadolinium.Reference Griffin, Delapaz and Euzmann14 A recent study showed that diffusion-weighted MRI has greater accuracy in detecting cholesteatoma.Reference Aarts, Rogers, van der Veen, Schilder, van der Heijden and Grolman15
A thorough assessment of congenital canal atresia patients is of the utmost importance when determining surgical candidacy for canalplasty. There are surgical risks and difficulties when approaching these patients. The main aim of canal surgery is hearing improvement, ideally a hearing level of 25 dB or better.Reference Declau, Cremers and Van de Heyning16, Reference Jahrsdoerfer, Yeakley, Aguilar, Cole and Gray17 Reconstruction of the external auditory canal is deferred until the age of five years, when the tympanic ring is fully developed and the course of the facial nerve is in its adult position. However, in cases with associated microtia, pinna reconstructive surgery supersedes canal surgery.
The criteria for surgical candidacy for canalplasty include a well developed middle-ear cavity and mastoid, with almost normal ossicles and a normal course of the facial nerve. The inner ear structures should also be normal, with no severe head or neck malformations. Jahrsdoerfer et al. have proposed a classification system to predict the prognosis following canal surgery.Reference Jahrsdoerfer, Yeakley, Aguilar, Cole and Gray17 According to their system, the best surgical candidates are those with scores of eight or more, while patients scoring five or less do poorly.
On the other hand, when congenital canal atresia patients also have cholesteatoma, surgical intervention must not be delayed. The extent of the cholesteatoma determines the surgical approach for these patients. Canalplasty is indicated when the cholesteatoma is confined to the external auditory canal. However, mastoidectomy is more appropriate for patients with extensive cholesteatoma involving the mastoid cavity.
In the current series, we found no relationship between the cholesteatoma extent and severity and the age of presentation. Patients undergoing canalplasty were aged between five and 27 years (mean age, 14.3 years), whilst those undergoing mastoidectomy were aged between four and 73 years (mean age, 26.7 years). This shows that patients can present with extensive cholesteatoma even at a young age, but also that, as expected, the disease tends to be more extensive in late presentations.
Close follow up is necessary to ensure the patency of the newly formed ear canal. In the early post-operative period, our patients were seen on a weekly or fortnightly basis. During these visits, adequate ear cleaning and management of granulation tissue or infection are crucial for a good surgical outcome. In our series, only two patients required repeated canal reconstruction surgery, due to soft tissue stenosis. Others had a good recovery, or required only skin grafting in the clinic. There was no recurrence of cholesteatoma.
In patients with canal atresia associated with cholesteatoma, hearing outcome is of secondary importance. The main objective of these patients' management is to ensure complete cholesteatoma removal, in order to prevent recurrence and complications. In the current series, a good post-operative hearing outcome (i.e. to within 30 dB) was achieved in five patients (38.5 per cent).
• Congenital abnormalities of the external auditory canal are uncommon
• Associated cholesteatoma must be suspected in all cases
• High resolution temporal bone computed tomography is mandatory at four years of age
• Recurrent otorrhoea is the commonest presenting symptom
• Patients can present much later in life with complications of cholesteatoma
Our centre's management protocol for congenital canal atresia includes high resolution CT scanning at four years of age in all cases.
In cases of bilateral canal atresia, early hearing amplification with bone conduction hearing aids is important to ensure normal speech development.
Subsequent management of congenital canal atresia patients will depend on the high resolution CT findings. The presence of cholesteatoma needs to be addressed urgently, before any cosmetic or functional surgery is considered. If there is no cholesteatoma present, the choice of surgery will depend on the status of the ossicles, the position of the facial nerve and the pneumatisation of the mastoid air cells. If the middle-ear status is favourable, canalplasty is the best surgical option. However, if the middle ear is poorly developed, a bone-anchored hearing aid is a better and safer choice.
Conclusion
Congenital external auditory canal atresia is a rare condition. One must always suspect the presence of underlying cholesteatoma in such cases. Cholesteatoma may be silent for many years, or may present with recurrent ear discharge or complications. High resolution CT of the temporal bones is advocated at four years of age to detect early cholesteatoma and to assess candidacy for reconstructive surgery. When cholesteatoma is present, surgical exploration is necessary to create a safe, dry ear, even at the expense of the hearing outcome.




