


Introduction
The Ewing's sarcoma family of tumours comprises a group of well characterised neoplasms, including Ewing's sarcoma of bone, extraskeletal Ewing's sarcoma, Askin tumour and malignant primitive neuroectodermal tumours.Reference West1,Reference Thacker, Temple and Scully2 These tumours are histologically similar, small, blue, round tumours, all of which show a characteristic 11q22q chromosomal translocation which results in the expression of a chimeric protein. The type of fusion transcript impacts on the clinical biology. These tumours occur in childhood and adolescence, with a median age at diagnosis of 15 years. They are aggressive neoplasms, with 25 per cent of patients having clinically evident metastases at presentation. Ewing's sarcoma has therefore been considered as a systemic disease, necessitating local as well as systemic treatment. Before the introduction of chemotherapy, nearly all patients died of their disease. However, the introduction of new chemotherapeutic agents has resulted in a major improvement in patient survival. Modern therapy consists of high dose chemotherapy for systemic control of disease, followed by local control, with wide surgical resection and/or irradiation. A few chemotherapeutic agents have been identified as active, such as vincristine, doxorubicin and cyclophosphamide. The integration of ifosfamide and etoposide, in conjunction with vincristine, doxorubicin and cyclophosphamide based therapy, has significantly improved response rates and survival.Reference Wexler, DeLaney, Tsokos, Avila, Steinberg and Weaver-McClure3
Extraskeletal Ewing's sarcoma, first reported by Tefft et al. in 1969,Reference Tefft, Vawter and Mitus4 is extremely rare in comparison with Ewing's sarcoma. The majority of cases have been reported in the lower limb and paravertebral region, with few cases reported in the neck.
We report a case of extraskeletal Ewing's sarcoma arising from the cervical spine, presenting as a neck mass. Characteristic features on computed tomography (CT) and magnetic resonance imaging (MRI) scans, in conjunction with histological analysis, were helpful in making the diagnosis.
Case report
A 14-year-old boy presented with a one-month history of a rapidly expanding, right-sided neck mass.
Examination showed a large, right neck mass deep to the sternocleidomastoid muscle, extending from the angle of the mandible to the superior border of the clavicle and measuring approximately 12 × 8 cm. Flexible endoscopy showed a submucosal mass filling the right hypopharynx and a right-sided vocal fold palsy. Neurological examination showed all other cranial nerves to be intact. Paraesthesia of the upper, medial aspect of the right arm was detected, indicating impairment of the T1 nerve root distribution of the brachial plexus.
Computed tomography demonstrated a 12 cm (craniocaudal) × 5 cm (transverse) mass on the right side of the neck, arising from the paravertebral space and bulging into the right carotid space, and displacing the midline viscera to the left, the right common carotid artery anteromedially and the internal jugular vein anterolaterally (Figure 1a). The mass was noted to be multiloculated, with the caudal aspect of the lesion appearing more solid. The vertebral artery was noted to pass directly through (and to be encased by) the caudal aspect of the mass (Figure 2). However, the artery opacified with contrast and was thus still patent, with no obvious compression.

Fig. 1 The multiloculated, ‘cystic’ nature of the tumour, as demonstrated by (a) low attenuation on axial computed tomography, and (b) high signal intensity on T2-weighted, axial magnetic resonance imaging. Arrow indicates anteromedial deviation of the carotid artery.

Fig. 2 (a) Axial computed tomography and (b) T1-weighted, axial magnetic resonance imaging, showing the vertebral artery passing through the tumour (arrow), and displacement of the midline viscera.
An MRI scan showed that the loculations displayed high signal intensity on T2-weighted images, intermediate signal on T1-weighted images and no enhancement post-contrast (but with some enhancement of the solid component). The MRI scan also showed that the tumour was infiltrating through the neural exit foramina of the lower cervical nerve roots, C6 to T1 (Figure 3). This was particularly well demonstrated at the level of the C7 nerve root, where low signal intensity tumour (on T1-weighted images) was demonstrated to obliterate fat within the exit foramen and to extend centrally towards the vertebral canal, but without any significant component within the vertebral canal itself (Figure 4).
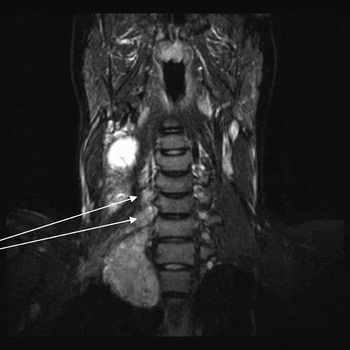
Fig. 3 T2-weighted, saggital magnetic resonance imaging, showing tumour extending through neural exit formen (arrows).

Fig. 4 T1-weighted, axial magnetic resonance imaging, showing right vertebral artery (top left arrow), tumour extending through neural exit foramen (bottom left arrow), the left vertebral artery (top right arrow) and lower cervical nerve root (bottom right arrow).
At this stage, a review of the CT scan did not demonstrate any bony widening or enlargement of the neural exit foramina, nor any evidence of bony destruction. Displacement of the longus colli muscle medially suggested that the lesion arose from the paravertebral space rather than the carotid space, as had been initially thought.
Based on the imaging findings alone, the initial diagnostic possibilities that were entertained included malignant peripheral nerve sheath tumour, rhabdomyosarcoma and neuroblastoma.
Biopsy (initially fine needle aspiration (FNA) and subsequently core biopsy) showed features suggestive of a sarcoma. Immunohistochemistry and chromosomal analysis showed the mass to be a Ewing's sarcoma. Since the mass was not arising within bone, the mass was diagnosed as an extraskeletal Ewing's sarcoma.
The management of the Ewing family of sarcomas is by multiagent chemotherapy for systemic control, and either wide surgical resection and post-operative radiotherapy or irradiation alone for local control. In this particular case, the patient was considered to have unresectable disease due to involvement of the cervical roots of C5–T1 and encasement of the vertebral artery. Any attempt at surgical debulking was also considered to be unacceptable, due to the risk of stroke from damage to the vertebral artery, and the risk of upper limb paralysis from damage to the cervical roots C5 to T1. The decision was therefore made to treat with combination chemotherapy for systemic control, followed by irradiation for local control. Before treatment, distant metastatic disease was excluded by bone marrow aspirate and trephine biopsy and also by imaging of the chest, abdomen and pelvis. (This was important, as the treatment response and subsequent prognosis is much poorer in the setting of distant metastatic disease.)
The combination chemotherapy regimen undertaken comprised six cycles of vincristine, ifosamide, doxorubicin and etoposide, administered three weeks apart. During the second cycle, the patient required admission for symptoms of pyrexia, vomiting and abdominal pain, attributed to febrile neutropenia and settled with conservative treatment. Following completion of the six cycles, further chemotherapy was given for a period of five weeks, comprising vincristine, adriamycin and cyclophosphamide. The patient then had a six-week course of external beam irradiation for local control.
Subsequent MRI showed a complete response to therapy. At the time of writing, the patient was being kept under close observation with three-monthly review.
Discussion
Extraskeletal Ewing's sarcoma was first described by Tefft et al. Reference Tefft, Vawter and Mitus4 in four patients with paravertebral soft tissue sarcomas. Angervall and EnzingerReference Angervall and Enzinger5 went on to describe the histological features of a series of 39 patients with soft tissue, round cell sarcomas considered histologically indistinguishable from Ewing's sarcoma of bone. Their series mainly involved young adults, in whom these tumours were usually found in the extremities and paravertebral region, with only one located around the cervical spine. These tumours were frequently rapid growing and displayed varying degrees of necrosis and degeneration, the latter features often accompanied by the finding of thick, fibrous septa and haemorrhage. Neurological symptoms or signs were a frequent concomitant. In a review of the radiological features of extraskeletal Ewing's sarcoma by O'Keefe et al., Reference O'Keefe, Lorigan and Wallace6 half were found to be located in the extremities and the other half in the chest, abdomen or pelvis. Within the chest, the majority were located in the paravertebral region. Although the imaging characteristics are non-specific, these masses tended to be well circumscribed (this feature being ascribed to the frequent pathological finding of a pseudocapsule), of generally low attenuation in relation to muscle on CT, and frequently hypoechoic on ultrasonography (said to reflect necrosis or cyst formation). Tumour vascularity was said to be variable. In our patient, the lesion was noted to be multiloculated, with an apparently ‘cystic’ appearance demonstrated as low attenuation on CT and high signal on T2-weighted images.
Extraskeletal Ewing's sarcoma presenting as a neck mass is extremely rare. A recent case reportReference Kennedy, Eustace, Caulfield, Fennelly, Hurson and O'Rourke7 described a rapidly enlarging, left posterior triangle neck mass in a 24-year-old. This patient, as in our case, demonstrated neurological signs in the ipsilateral upper limb. Interestingly, as with our case, this mass was reported to infiltrate through a number of intervertebral foramina and to encase a segment of the vertebral artery. No bony abnormality was found. More recently still, the case of a nine-year-old boy presenting with neck pain and right arm weakness has been described.Reference Hardasmalani, Naim, Kroning and Bithoney8 Cervical CT revealed a right-sided, paraspinal mass, and MRI demonstrated a significant epidural component and extension through a number of neural foramina. The occurrence of neurological symptoms in this condition has also been highlighted in a report of two further cases,Reference Modrego and Pina9 one occurring in the lumbosacral region in a 16-year-old, another occurring, unusually, in the cervical region of a 74-year-old. Again, involvement of neural exit foramina was a feature.
• Extraskeletal Ewing's sarcoma is part of the Ewing's sarcoma family of tumours characterised by 11q22q chromosomal translocation
• Extraskeletal Ewing's sarcoma has a propensity to occur in a paraverterebral location and is found rarely in the cervical region
• Extraskeletal Ewing's sarcoma has a predilection to infiltrate neural exit foramina and to encase vascular structures
• Therefore, both magnetic resonance and computed tomography imaging are useful in making the diagnosis
• Treatment requires multiagent chemotherapy for systemic control, and surgery and/or irradiation for local control
• Cure rates of 70 per cent have been reported for localised disease. The cure rate is 30 per cent if patients have metastatic disease at presentation
• Extraskeletal Ewing's sarcoma has a poorer outcome than Ewing's sarcoma
Modern therapy for extraskeletal Ewing's sarcoma is the same as for all the Ewing family of sarcomas.Reference Carvajal and Meyers10 This consists of high dose chemotherapy for systemic control of the disease, followed by local control, either with wide surgical resection and post-operative radiotherapy or with irradiation alone. Due to the rarity of extraskeletal Ewing's sarcoma, survival figures for this condition are reported as a group with the rest of the Ewing family of sarcomas. For patients with localised disease, cure rates of 70 per cent can be achieved.Reference Longhi, Setola, Versari and Bacci11 However, if patients have metastatic disease at presentation, the cure rate is less than 30 per cent.Reference Longhi, Setola, Versari and Bacci11 The choice of surgical resection or irradiation for local control is dependent on surgical resectability and morbidity. However, there is some evidence that wide surgical resection and post-operative irradiation produces better local control than irradiation alone, particularly for patients with Ewing's sarcoma of the extremity.Reference Bacci, Longhi, Briccoli, Bertoni, Versari and Picci12 BacciReference Bacci, Longhi, Briccoli, Bertoni, Versari and Picci12 reported a local control rate of 88 per cent for surgery and post-operative irradiation, versus 80 per cent for irradiation alone (p < 0.009). For systemic control, several different chemotherapy regimes can be used, including vincristine, doxorubicin, cyclophosphamide, ifosfamide and etoposide,Reference La, Meyeres, Wexler, Alektiar, Healey and Laquaglia13 and vincristine, doxorubicin, cyclophosphamide and actinomycin D.Reference Krasin, Davidoff, Rodriquez-Galindo, Billups, Fuller and Neel14 Using the former regime for systemic control and irradiation for local control, La et al. Reference La, Meyeres, Wexler, Alektiar, Healey and Laquaglia13 reported a three-year overall survival rate of 86 per cent for patients with localised disease and 21 per cent for patients with metastatic disease at presentation. Using vincristine, doxorubicin, cyclophosphamide and actinomycin D for systemic control and surgery for local control, Krasin et al. Reference Krasin, Davidoff, Rodriquez-Galindo, Billups, Fuller and Neel14 reported an overall five-year survival rate of 84 per cent and a local recurrence rate of 12.5 per cent. The site of tumour was a significant predictor of survival, with extraskeletal Ewing's sarcoma having a poorer outcome. For patients who have recurrent disease the outcome is poor, although specific subgroups can be cured even after two or three relapses (i.e. patients who relapse two or more years after primary treatment; patients who relapse with only lung metastases; and patients whose recurrences can be surgically treated).Reference Bacci, Longhi, Ferrari, Mercuri, Barbieri and Bertoni15 However, McTiernan et al. Reference McTiernan, Driver, Michelagnoli, Kilby and Whelan16 has reported a five-year survival rate of 38 per cent in patients treated with peripheral bone marrow transplant followed by busulphan and melphalan therapy.
Conclusion
We report a case of extraskeletal Ewing's sarcoma presenting as a neck mass. The case is discussed in relation to the literature. It is noted that extraskeletal Ewing's sarcoma has a propensity to occur in a paravertebral location, being found rarely in the cervical region. We also note that this neoplasm seems to have a predilection to infiltrate through neural exit formina (without necessarily invading bone) and to encase vascular structures such as the vertebral artery. Although the imaging features are of themselves non-specific, this rare entity should be considered in the differential diagnosis of patients with similar presentations.




