


Introduction
Sparganosis is an important neglected parasitic disease caused by invasion by spargana, plerocercoid larvae of various diphyllobothroid cestodes belonging to the genus Spirometra. Sparganosis is a zoonosis from amphibians, reptiles or mammals causing significant economic losses and also a public health problem in humans (Ooi et al., Reference Ooi, Chang, Huang, Kawakami and Uchida2000; Pampiglione et al., Reference Pampiglione, Fioravanti and Rivasi2003; Wiwanitkit, Reference Wiwanitkit2005). Although human sparganosis is a worldwide parasitic zoonosis, it is most frequently found in eastern and south-eastern Asia, including China (Cui et al., Reference Cui, Lin, Zhang, Xu and Wang2011). The spargana invade mainly the brain, eye, spinal cord, subcutaneous tissues and abdominal cavity, and can cause blindness, paralysis and even death (Li et al., Reference Li, Lin, Xie, Gao, Huang, Wu, Li, Lin and Zhu2009). Human infections are mainly acquired by eating raw or insufficiently cooked meat of frogs and snakes.
A traditional approach for the identification of Spirometra is by morphological observation, but this approach has limitations in the differentiation of Spirometra erinaceieuropaei from other similar species such as Spirometra manson, therefore causing confusion. However, accurate identification of parasites and their characterization at different taxonomic levels have important implications for the prevention and control of parasitic diseases. A range of studies has demonstrated that molecular markers are of particular utility for species-specific identification and detection of a number of parasite groups (Sun et al., Reference Sun, Zhu, Xie, Wu, Li, Lin and Song2006, Reference Sun, Noe, Barber, Coyne, Cassidy-Hanley, Clark, Findly and Dickerson2009).
Sequence variation is widespread in parasite populations, and the accurate analysis of genetic variation in parasites has important implications for the genetic structure of parasites, epidemiology and studying population biology. Previous studies have shown that the cytochrome c oxidase subunit 3 gene (cox3) is the preferred gene for genetic variation and phylogentic analyses, due to sufficient variability and a consistent phylogenetic signal (Zarowiecki et al., Reference Zarowiecki, Huyse and Littlewood2007; Zhao et al., Reference Zhao, Mo, Zou, Weng, Lin, Xia and Zhu2009), and NADH dehydrogenase subunits 1 and 4 genes (nad1 and nad4) also have more characters of phylogenetic information and variability (Gasser et al., Reference Gasser, Zhu and McManus1999; Zhao et al., Reference Zhao, Mo, Zou, Weng, Lin, Xia and Zhu2009). Therefore, they provide better markers for both phylogenetic and population studies. However, there is a paucity of information on the genetic variation in populations of some important parasite groups from China, such as S. erinaceieuropaei spargana of human and animal health significance.
The objectives of the present study were to examine sequence variability in mitochondrial cox3, nad1 and nad4 regions, among S. erinaceieuropaei spargana isolates from different endemic regions in China. Based on the pcox3, pnad1 and pnad4 sequences, the phylogenetic relationships of S. erinaceieuropaei spargana were also reconstructed.
Materials and methods
Parasites and isolation of genomic DNA
The parasite species, with their sample codes, number of samples, host species and geographical origins are listed in table 1. Total genomic DNA was extracted from individual samples by sodium dodecyl sulphate/proteinase K treatment, column-purified (Wizard™ DNA Clean-Up, Promega, Madison, Wisconsin, USA) and eluted into 50 μl water according to the manufacturer's recommendations.
Table 1 Geographical origins (different locations in Hunan province) of S. erinaceieuropaei spargana samples used in the present study, as well as their GenBank™ accession numbers for sequences of partial mitochondrial cytochrome c oxidase subunit 3 gene (pcox3), NADH dehydrogenase subunits 1 and 4 genes (pnad1 and pnad4).

Enzymatic amplification
The primer sets for cox3, nad1 and nad4 genes were designed by the authors, based on well-conserved sequences in many distantly related taxa (table 2). These primers were synthesized on a Biosearch Model 8700 DNA synthesizer (Shanghai, China). Polymerase chain reactions (PCRs) (25 μl) were performed in 2 mm MgCl2, 2.5 μm of each primer, 2.5 μl 10 × rTaq buffer, 0.2 mm of each deoxyribonucleoside triphosphate (dNTP), 1.25 U of rTaq DNA polymerase (Takara, Dalian, China), and 1 μl of DNA sample in a thermocycler (Biometra, Göttingen, Germany) under the following conditions: after an initial denaturation at 94°C for 5 min, then 94°C for 30 s (denaturation); 55°C (for pcox3, pnad1 and pnad4) for 30 s (annealing); 72°C for 30 s (extension) for 38 cycles, followed by a final extension at 72°C for 10 min. These optimized amplification conditions for the specific and efficient amplification of individual DNA fragments were obtained after varying annealing and extension temperatures. One microlitre (5–10 ng) of genomic DNA was added to each PCR reaction. Samples without genomic DNA (no-DNA controls) were included in each amplification run, and in no case were amplicons detected in the no-DNA controls (not shown). Five microlitres of each amplicon was examined by 0.8% (w/v) agarose gel electrophoresis to validate amplification efficiency. PCR products were sent to Sangon Company (Shanghai, China) for sequencing using a primer walking strategy.
Table 2 Sequences of primers used to amplify a portion of the mitochondrial cytochrome c oxidase subunit 3 gene (pcox3), NADH dehydrogenase subunits 1 and 4 genes (pnad1 and pnad4) from S. erinaceieuropaei spargana in the present study.

Sequence analysis and phylogenetic reconstruction
Sequences of the three mitochondrial genes were separately aligned using the computer program Clustal X 1.83 (Thompson et al., Reference Thompson, Gibson, Plewniak, Jeanmougin and Higgin1997). Pairwise comparisons were made of the level of sequence differences (D) among and within the species using the formula D = 1 − (M/L), where M is the number of alignment positions at which the two sequences have a base in common, and L is the total number of alignment positions over which the two sequences are compared (Chilton et al., Reference Chilton, Gasser and Beveridge1995).
Phylogenetic analyses were based on the sequences of the three mitochondrial genes available in this study. Three methods, namely neighbour joining (NJ), maximum likelihood (ML) and maximum parsimony (MP), were used for phylogenetic reconstructions. Standard unweighted MP was performed using the package Phylip 3.67 (Felsenstein, Reference Felsenstein1995). NJ analysis was carried out using the Dayhoff matrix model implemented by MEGA 4.0 (Tamura et al., Reference Tamura, Dudley, Nei and Kumar2007), and ML analysis was performed using PUZZLE 4.1 under the default setting (Strimmer & Haeseler, Reference Strimmer and Haeseler1996). The consensus tree was obtained after bootstrap analysis, with 1000 replications for NJ and MP trees, and 100 for the ML tree, with values above 50% reported. To study the genetic relationships among diphyllobothroid cestodes, other members of the Diphyllobothriidae were considered in the present study (Diphyllobothrium nihonkaiense NC_009463; D. latum AB269325; Spirometra erinaceieuropaei NC_011037), with Taenia solium (GenBank™ accession number NC_004022) as the outgroup. Phylograms were drawn using the Tree View program version 1.65 (Page, 1996).
Results and discussion
Genomic DNA was extracted from 33 individual spargana representing 11 geographical locations in Hunan Province, China (fig. 1). pcox3, pnad1 and pnad4 (~600, 650 and 900 bp, respectively) were amplified individually and subjected to agarose gel electrophoresis. The results showed that no size variation was detected on agarose gels among any of the amplicons examined for each mtDNA region. To assess sequence variation in these three mtDNA regions within and between isolates, amplicons of pcox3, pnad1 and pnad4 from samples representing different isolates were selected and then subjected to sequencing. The sequences of pcox3, pnad1 and pnad4 were 541, 607 and 847 bp in length, respectively. The A+T contents of the sequences were 68.39–68.76% (pcox3), 63.76–64.91% (pnad1) and 67.18–67.77% (pnad4), respectively, while the intra-specific sequence variations within each of the S. erinaceieuropaei spargana were 0–1.5% for pcox3, 0–2.8% for pnad1 and 0–2.7% for pnad4, consistent with a recent report by Liu et al. (Reference Liu, Zhao, Tan, Zeng, Wang, Yuan, Lin, Zhu and Liu2010). For pcox3, no sequence variation was detected among Changsha, Hengyang, Xiangtan and Zhangjiajie isolates; while sequence variations of more than 0.5 were found among Xupu, Yiyang and Yueyang isolates. For pnad1, sequence variations of more than 3.0 were detected in four locations, including Chenzhou, Loudi, Xupu and Yeyang isolates. For pnad4, sequence variations of more than 2.0 were found among Xupu and Yueyang isolates.

Fig. 1 The sampling locations for S. erinaceieuropaei spargana isolates in different regions of Hunan, China.
For pcox3 and pnad4, intra-specific nucleotide variation was related mainly to changes at the third codon position, while fewer changes were detected at the first or second codon positions (table 3), consistent with results of other organisms (Li et al., Reference Li, Lin, Song, Sani, Wu and Zhu2008; Zhao et al., Reference Zhao, Mo, Zou, Weng, Lin, Xia and Zhu2009). For example, the number of intra-specific variations were 4, 0, 7 for the first, second and third positions for cox1; 1, 4, 14 for nad1; and 8, 1, 15 for nad4, respectively. Intra–specific nucleotide variations represented transitions (A ↔ G or C ↔ T; n = 10 for pcox3, n = 17 for pnad1 and n = 19 for pnad4), transversions (A ↔ C, A ↔ T, C ↔ G, and/or T ↔ G; n = 1 for pcox3, n = 2 for pnad1, n = 5 for pnad4).
Table 3 Number and position of codon variations in a portion of the mitochondrial cytochrome c oxidase subunit 3 gene (pcox3) and NADH dehydrogenase subunits 1 and 4 genes (pnad1 and pnad4) among S. erinaceieuropaei spargana isolates in China.
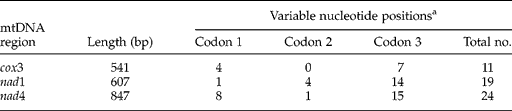
a The first codon position of each sequence was determined in relation to the complete mitochondrial DNA sequence of S. erinaceieuropaei (NC_011037).
The combined sequences of pcox3, pnad1 and pnad4 representing different isolates were aligned over a consensus length of 1853 bp. Topologies of all trees constructed by different methods (NJ, MP and ML) with different building strategies and/or different distance models were identical or similar, with only small difference of bootstrap values (fig. 2). These results indicate that all the spargana isolates in Hunan Province represent S. erinaceieuropaei. From the phylogenetic tree: parasites of genus Diphyllobothrium were sister to the genus Spirometra, and S. erinaceieuropaei and D. nihonkaiense were more closely related to the other members of the Diphyllobothrium genus (D. latum), consistent with results of previous classifications based upon cox1 datasets (Liu et al., Reference Liu, Lin, Li, Liu, Liu, Yuan, Song, Zhao, Zhang and Zhu2011).

Fig. 2 Phylogenetic relationship among the examined cestode species inferred by maximum parsimony (MP), maximum likelihood (ML) and neighbour joining (NJ) analyses based on combined mitochondrial dataset (pcox3+ pnad1+ pnad4) sequences, using one Taenia species (Taenia solium) as outgroup. The numbers along branches indicate bootstrap values resulting from different analyses in the order: MP/ML /NJ. Values lower than 50 are given as ‘Weak’.
In conclusion, the genetic variability among S. erinaceieuropaei spargana isolates from different endemic regions in China could be revealed by sequences of three mitochondrial DNA genes. For the three mtDNA genes, genetic variation of pnad1 was higher than pnad4 and pcox3, and pnad4 was higher than pcox3. The results of the present study also have implications for the diagnosis and control of S. erinaceieuropaei spargana infections of animal and human health significance.
Acknowledgements
Project support was provided in part by the Department of Science and Technology, Hunan Province (Grant No. 2010FJ3006), the National Natural Science Foundation of China (Grant No. 30771616), and general project of Changsha Science and Technology plan (Grant No. K0902144-21).





