



Introduction
Early life adverse events can contribute to disease later in life, but not all individuals are affected to the same extent. These differences can be partially attributed to interactions between genetic variation and environmental risk factors such as maternal nutrition.Reference Godfrey, Reynolds and Prescott 1 – Reference Ong, Lin and Holbrook 3 Investigating these gene by environment interactions can improve our understanding of non-communicable disease risk. This can be achieved by moving to a systems-wide view of the processes that are required to decode the information (e.g. genes) that is encoded within the linear sequence of the DNA. In effect, we must combine genomic and post-genomic approaches to interpret genome biology so that we can understand how developmental processes are affected by the combinatorial action of genetic variation and epigenetics. Here we will discuss recent attempts to link genetic risk factors to environmental responses and disease risk through the incorporation of the three-dimensional organization of the genome.
Genes are supervened on the genome organization
What is the nature of the information within the DNA sequence? Genes are an obvious candidate. Yet, the view that a gene is hard-coded in the DNA sequenceReference Carlson 4 – Reference Gerstein, Bruce and Rozowsky 7 has a number of limitations. Notably, it is clear that genes are not fixed entities; rather they are supervened on the genome in a manner which is context dependent and programmable by the environment.Reference Lamm 8 This is supported by observations that the functions of defined DNA sequences are context dependent.Reference Griffiths and Neumann-Held 9 For example, a promoter may become part of an intron resulting in production of a chimeric messenger RNA transcribed from groups of exons that were previously ascribed to different genes.Reference Akiva, Toporik and Edelheit 10 If one extends the definition of the gene to include the sequences that regulate transcription, then current evidence demonstrates that these elements are not fixed, nor necessarily in cis within the linear DNA sequence. Rather, the combinations are cell-type specific and this is reflected in the spatial organization of the DNA.Reference Spilianakis, Lalioti and Town 11 – Reference Rao, Huntley and Durand 15
Genome organization: a definition
When looking at a static microscopic image of a nucleus it is easy to forget that it is in a state of non-equilibrium, constantly exchanging its material constituents with the cytoplasm.Reference Bischof 16 This non-equilibrium is most elegantly demonstrated by the formation of condensed chromosomes from interphase DNA as the cell enters metaphase of the cell cycle. Yet the DNA is spatially ordered within the nucleus throughout all phases of the cell cycle; chromosomes reside in regular domains within the nucleus known as chromosome territories. As such, the three-dimensional organization of a genome should be thought of as an emergent property of that particular genome in the context of the micro- (i.e. nuclear, intra-cellular) and macro-environments (inter- and extra-cellular) to which that genome is exposed. Notably, within a population absolute structure cannot be achieved, as there will always be a degree of stochasticity between the genome structure in identical cells exposed to identical conditions as a result of diffusion of molecules and random movement of loci (Brownian motion).Reference O’Sullivan, Hendy and Pichugina 17 Nonetheless, if we capture the genome structure at any one moment in a particular cell, by definition it must have a single structure.
Proximity ligation and modern microscopic approaches are capable of capturing genomes in the different spatial organizations that they assume. Despite the inherent limitations of these methods,Reference Grand, Gehlen and O’Sullivan 18 results from recent studies suggest that the genome and nucleus collectively forms a constrained system that is maintained on the boundary of order and chaos.Reference Kauffman 19 Within this constrained system, genomes are interleaved entitiesReference Kapranov, Willingham and Gingeras 20 that are spatially organized into hierarchically organized domains of different sizes (e.g. chromosome territories and topological associated domains).Reference Fraser, Ferrai and Chiariello 14 , Reference Dixon, Selvaraj and Yue 21 The organization of these domains enables the rapid, simultaneous and appropriate accessing of hard-coded information within the DNA sequence as chromatin regions come in and out of contact.
Reproducible and directed changes to genome organization are observed throughout the cell cycleReference Grand, Gehlen and O’Sullivan 18 and development.Reference Dixon, Jung and Selvaraj 12 , Reference Rao, Huntley and Durand 15 , Reference de Wit, Bouwman and Zhu 22 , Reference Krijger, Di Stefano and de Wit 23 For example, reprogramming of mouse pre-B cells, bone-marrow derived macrophages, neural stem cells and embryonic fibroblasts demonstrated that early passage induced pluripotent stem cells carry reproducibly acquired features of genome organization that are contingent on their cell of origin.Reference Krijger, Di Stefano and de Wit 23 Assuming that genome organization emerges from the positioning of chromatin (Fig. 1), it is likely that metastable genome conformations are captured by the combined effects of environmentally signalled changes to the synthesis and degradationReference Buckley, Aranda-Orgilles and Strikoudis 31 of proteins and RNAReference Kim, Marinov and Pepke 32 that occur during the reprogramming. These programmes of change are dependent upon the cell-of-origin composition of transcription factors, proteins and RNAs, and the environmental signals that the cell is exposed to. In such a scenario, genome organization is not deterministic. Rather, it captures the sum activity of the nuclear functions that are occurring at a moment in time, including patterns of gene regulationReference Grand, Pichugina and Gehlen 33 – Reference Mifsud, Tavares-Cadete and Young 37 and ultimately cell fate choices.Reference Spilianakis, Lalioti and Town 11 , Reference Williams, Spilianakis and Flavell 38 These choices often occur in early development, but can affect the activity of key metabolic organs for a lifetime.Reference Felipe Barella, ulio Cezar de Oliveira and Cezar de Freitas Mathias 39 , Reference Vickers 40
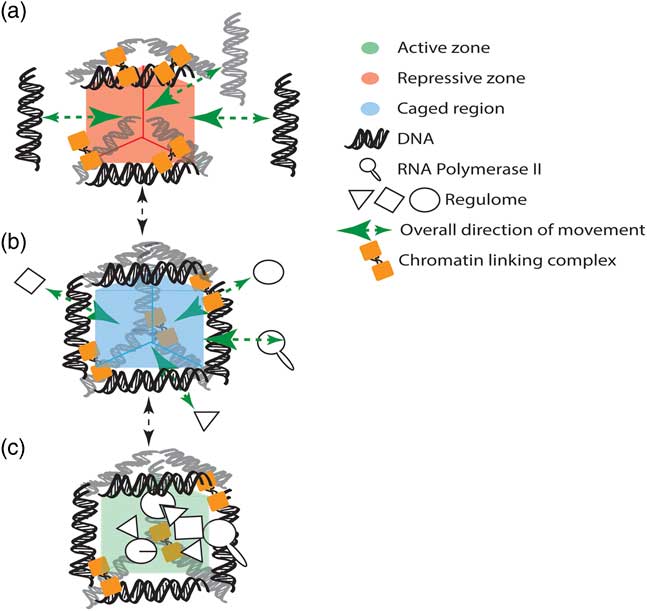
Fig. 1 Genomic structure emerges from the positioning of chromatin by either active or passive means to create phase separated subcompartments for stable gene regulation, repair and replication. (a) Chromatin is held in position by complexes (e.g. CTCF and cohesinReference Holwerda and de Laat 24 – Reference Mizuguchi, Fudenberg and Mehta 26 ), which are continuously binding and releasing the DNA template. (b) The structured chromatin creates a region in which diffusible nuclear components become retarded (i.e. caged region). (c) Concentrations that effect phase transitions and promote nuclear functions are ultimately attained.Reference Brangwynne, Tompa and Pappu 27 In this model, the retention within the caged region is promoted by high numbers of binding sites directly in the co-located chromatin loci or with other proteins bound to the chromatin.Reference Kampmann 28 – Reference Erdel, Müller-Ott and Rippe 30
How does genome structure link to the developmental origins of disease?
Metabolic disorders such as obesity and diabetes are recognized as being highly heritable, but despite significant progressReference Jarick, Vogel and Scherag 41 – Reference Sladek and Prokopenko 48 their genetic basis has not been fully explained.Reference Manolio, Collins and Cox 49 , Reference Vattikuti, Guo and Chow 50 The majority of disease-associated single-nucleotide polymorphisms (SNPs) (daSNPs) are found in non-coding regions of the genome.Reference Farh, Marson and Zhu 51 Traditionally, these intergenic or intronic daSNPs have been thought to act on the nearest gene, under the assumption that regulatory interactions involve cis acting sequences that are linked, or proximal, to the gene of interest.Reference Schierding, Cutfield and O’Sullivan 52 Although this assumption is often correct, the three-dimensional nature of the genome allows regulatory sequences to interact with and modify the expression of distal genes; these may be many kilobases (kb) or megabases away on the same chromosome, or even on different chromosomes.Reference Spilianakis, Lalioti and Town 11 , Reference Marsman and Horsfield 53 , Reference Sanyal, Lajoie, Jain and Dekker 54
Although the exons of a gene tend to occur in a linear order along the chromosome, the DNA elements that are necessary for the regulation of gene transcription can be located almost anywhere within the genome.Reference Williams, Spilianakis and Flavell 38 , Reference Marsman and Horsfield 53 This includes distal intergenic regionsReference Chen and Tian 55 , Reference Schierding, Antony and Cutfield 56 and the introns of other genes.Reference Smemo, Tena and Kim 57 , Reference Claussnitzer, Dankel and Kim 58 However, in order to contribute to the regulation of gene expression, at least a subset of these regulatory elements must physically associate with the target gene promoter. This is facilitated by the formation of DNA loops which allow the element to come into spatial proximity with the target gene.Reference Tolhuis, Palstra and Splinter 59 , Reference Drissen, Palstra and Gillemans 60 A mutation in an enhancer element may disrupt this regulatory cluster, altering transcription of the target gene. Genetic variants that alter gene expression in this way are known as expression quantitative trait loci (eQTLs).Reference Albert and Kruglyak 61
eQTL analysis has proved valuable in assigning function to intergenic SNPs associated with disease in genome-wide association studies (GWAS).Reference Albert and Kruglyak 61 Combining eQTL analyses with chromatin capture techniques [e.g. chromosome conformation capture,Reference Naumova, Smith, Zhan and Dekker 62 circular chromosome conformation capture,Reference Zhao, Tavoosidana and Sjölinder 63 genome conformation capture,Reference Rodley, Bertels, Jones and O’Sullivan 64 high-throughput chromosome conformation capture (Hi-C)Reference Rao, Huntley and Durand 15 ], which detect spatial proximity of chromosomal loci, provides further evidence that an enhancer in which a SNP resides is spatially and functionally linked to the target gene.Reference Sanyal, Lajoie, Jain and Dekker 54 , Reference Schierding, Antony and Cutfield 56 , Reference Smemo, Tena and Kim 57 , Reference Schierding and O’Sullivan 65 – Reference Harmston and Lenhard 67 Utilizing spatial proximity data to identify candidate regulatory targets increases the power of the study; fewer putative eQTLs are calculated and thus the statistical correction for multiple testingReference Doss 68 , Reference Davis, Fresard and Knowles 69 is less severe.Reference Schierding, Antony and Cutfield 56 For example, an obesity-associated locus on chromosome 16, identified from GWAS studies, was found to have no effect on transcript levels of the nearest gene (FTO).Reference Smemo, Tena and Kim 57 Instead circular chromatin conformation capture followed by high-throughput sequencing (4C-seq) identified IRX3, a gene 300 kb away, as the target of the daSNPs.Reference Smemo, Tena and Kim 57 , Reference Claussnitzer, Dankel and Kim 58 These combined analyses help to interpret the effects of intergenic and non-coding SNPs by identifying the genes and genetic pathways that they affect. However, this approach relies upon the underlying assumption that intergenic and intronic daSNPs mark regulatory loci (e.g. enhancers, repressors, or modifiers of the aforementioned).
Intergenic SNPs are difficult to categorize, as they often fall outside conserved regions, non-coding RNAs, known enhancers, or distal regulatory elements. Chen and TianReference Chen and Tian 55 approached this issue by grouping all intergenic SNPs with their nearest regulatory element. They then predicted the target genes of each regulatory element using spatial proximity, epigenetic data and phylogenetic profiles.Reference Chen and Tian 55 This approach found that the predicted targets of the regulatory elements were often enriched for protein-coding genes associated with the investigated diseases. However, assigning SNPs to the closest regulatory element in cis, without evidence for a functional connection is a problematic assumption. In many respects this approach perpetuates our earlier practice of assigning SNPs to the closest protein-coding gene.
Combining information on the spatial organization and functional impact (e.g. eQTLs) of daSNPs to determine how they contribute to a phenotype is further complicated by the complexity of the regulatory circuits that exist within eukaryotic nuclei. For example, enhancers or repressors need not act individually. Rather, the elements are combinatorial and the tissue-specific manner in which they connect contributes to counteract stochastic variation in the regulation of the target gene. Consistent with this, Corradin et al. Reference Corradin, Cohen and Luppino 70 found that within clusters of super-enhancers, isolated SNPs can have large effects on the disease risk in combination with known risk SNPs, even if one variant does not reach genome-wide significance or have a detectable spatial interaction with the target gene. Moreover, variants that alter epigenetic patterns can affect not just local gene regulation but large scale genome organization. For instance the CCCTC-binding factor (CTCF) is a key architectural protein,Reference Ong and Corces 71 holding together megabase scale regions of DNA.Reference Nora, Goloborodko and Valton 72 These structures are known as topologically associated domains (TADs). It thought that TADs function to increase the incidence of contacts between loci within the TAD while simultaneously insulating genes in one TAD from the effects of enhancers in another.Reference Nora, Goloborodko and Valton 72 CTCF binding varies greatly between cell types, and can be sensitive to DNA methylation.Reference Wang, Maurano and Qu 73 , Reference Maurano, Wang and John 74 Variants that affect methylation patterns (meQTLs)Reference Banovich, Lan and McVicker 75 could therefore cause widespread transcriptional changes by disrupting TAD boundaries.Reference Flavahan, Drier and Liau 76
Future directions
Genome organization is a record of nuclear activity including gene regulation patterns.Reference de Wit, Bouwman and Zhu 22 , Reference Sanyal, Lajoie, Jain and Dekker 54 These marks can be used to further our understanding of phenotypes. For example, genome organization informed-discovery of allele-specific enhancer, insulator or promoter activity using intergenic SNPs can be integrated into GWAS to help explain the environment-genotype component of missing human heritability.Reference Schierding, Cutfield and O’Sullivan 52 However, accurate deconvolution of the nuclear activity requires accurate maps and contact-informed models of the genomic organization of different cell-types or tissues at different developmental or disease stages. The commonly used Hi-C technique requires hundreds of millions of reads in order to capture a representation of the interactions that are occurring in the genome.Reference Rao, Huntley and Durand 15 However, due to the complexity of these libraries, specific interactions are rarely sequenced to a sufficient depth for interrogation.Reference Mifsud, Tavares-Cadete and Young 37 Capture Hi-C is a method that enriches a Hi-C library for all interactions with, for example, gene promotersReference Mifsud, Tavares-Cadete and Young 37 or GWAS loci.Reference Jäger, Migliorini and Henrion 36 , Reference Martin, McGovern and Orozco 77 Use of this targeted approach enables the identification of all possible targets of non-coding risk loci identified by GWAS whilst overcoming limitations that are inherent to both microscopy and proximity ligation.Reference Grand, Gehlen and O’Sullivan 18 , Reference Dekker 78 , Reference de Wit and de Laat 79
A further limitation of both GWAS and Hi-C is that of resolution. GWAS can identify daSNPs, but they merely mark a locus that has potential regulatory effects associated with the phenotype of interest. The daSNP is typically in high linkage with one or more SNPs that are located within a linkage disequilibrium block. Similarly, Hi-C identifies an interacting region containing the tag SNP. However, linkage disequilibrium blocks can potentially cross several restriction fragments. Therefore, targeted methods such as Capture Hi-C must identify interactions that occur within the linkage disequilibrium block associated with the tag SNP – not simply the tag SNP itself.
It is currently not possible to bioinformatically determine the causal SNP within a region, but functional annotation can be used to prioritize SNPs for experimental follow-up.Reference Tak and Farnham 80 The patterns of enhancers, methylation, histone modification, protein binding sequences and DNase hypersensitivity sites can all be used to predict plausible causal SNPs using large, publicly available datasets.Reference Farh, Marson and Zhu 51 , Reference Claussnitzer, Dankel and Kim 58 , Reference Kichaev, Yang and Lindstrom 81 , Reference Pasaniuc and Price 82 Information about the spatial organization of the genome can also contribute to this prediction, particularly if multiple restriction enzymes were used during proximity ligation, reducing the fragment size and identifying the interacting region with greater precision (Fig. 2). These predictions should then be tested using gene editing techniques, such as CRISPR/Cas9,Reference Claussnitzer, Dankel and Kim 58 which enable the isolation of a specific SNP effect without losing the three-dimensional context of the interaction. Cell choice is essential in these types of study, due to the tissue specific nature of the genome organization.Reference Spilianakis, Lalioti and Town 11 – Reference Rao, Huntley and Durand 15
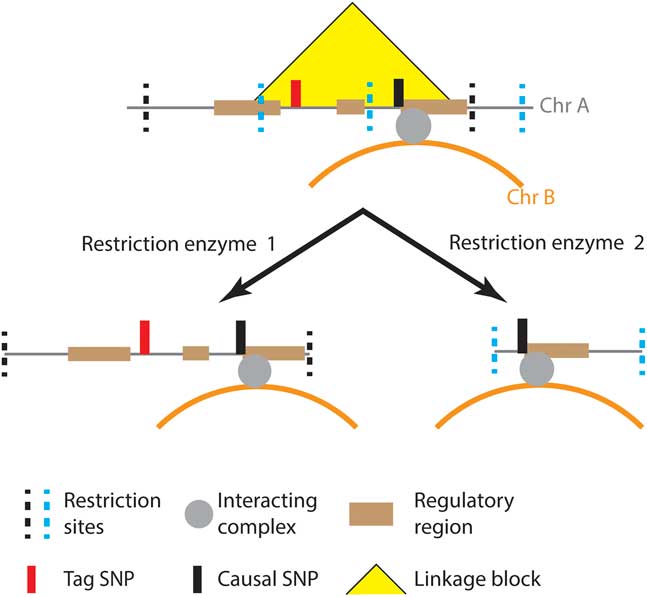
Fig. 2 Disease associated single-nucleotide polymorphisms (SNPs) identified by genome-wide association studies are often in linkage with one or more SNPs, any of which may be causal.Reference Farh, Marson and Zhu 51 Functional information, such as epigenetic marks and experimentally validated enhancers, is used to identify regulatory regions which are more likely to contain causal SNPs.Reference Claussnitzer, Dankel and Kim 58 , Reference Kichaev, Yang and Lindstrom 81 Comparisons of genomic organizations captured by proximity ligation with different restriction enzymes can be used to refine the identification of the interacting regions and the causal SNP.
Furthermore, carefully designed studies are required to find variants that increase disease risk only under specific environmental conditions,Reference Huang, Cate and Battistuzzi 83 or variants that may contribute to a pathogenic environment such as hyperphagia.Reference Yilmaz, Davis and Loxton 84
In multi-cellular organisms the nucleus is not a closed system and the genome is not a single entity. For example, interactions between the mitochondrial and nuclear genomes have been captured and linked to the control of gene expression, DNA repair and the cell cycle.Reference Rodley, Grand and Gehlen 85 , Reference Doynova, Berretta and Jones 86 Therefore, inter-organelle DNA interactions likely form a highly specific component of intra-cellular communication. Future work should investigate the potential for inter-organelle DNA interactions to contribute directly to the regulatory mechanisms through which daSNPs located in the mitochondria, and other nucleated organelles, contribute to complex phenotypes.
Conclusion
Gene regulation and regulatory networks are a critical component of developmental processes and environmental responses. Genome structure acts in a read–write capacity capable of capturing the underlying action of the regulome or possibly even directly inducing changes under conditions of physical stress.Reference Jacobson, Perry and Long 87 These interactions contribute to explaining how the various levels of nuclear control (structural, epigenetic and proteomic) come together to define genes and ensure cellular adaptation and selection through appropriate gene regulation, recombination and replication. Approaching the study of daSNPs from this viewpoint enables the interrogation of the genome as a complex organReference Lamm 88 capable of permutations to define genes in response to environmental stimuli. Including information about the distribution and dynamic profiles of other epigenetic marks can further increase the power of these analyses by identifying the effects of gene by environment interactions on the epigenome. Further work to describe the interleaved genome promises to elucidate how epigenetics contributes to the control of developmental pathways.Reference Bard 89
Acknowledgements
The authors would like to thank Phillip Smith, William Schierding and Tayaza Fadason for comments on this manuscript.
Financial Support
This work was supported by the Health Research Council New Zealand (grant number HRC 15/504 to JOS) and a University of Auckland Scholarship to E.J.
Conflicts of Interest
None.


