


Introduction
The classical view of mammalian development proposes that, other than to mediate aspects of androgenization of the male brain in some mammals, estrogens play an inconsequential role during organogenesis of the reproductive system and mammary glands. It was also assumed that, in the absence of male hormones, the default outcome would be feminine. Recent findings in the field of environmental endocrine disruption have revealed this view to be incorrect because developmental exposure to estrogenic chemicals has been shown to induce morphological, functional and behavioral anomalies associated with reproduction.Reference Markey, Coombs, Sonnenschein and Soto1–Reference Rubin, Murray, Damassa, King and Soto5 These and previous findingsReference Herbst and Bern6–Reference Boylan and Calhoon8 are prompting a re-evaluation of the basic concepts that drive developmental and cancer research.Reference Soto and Sonnenschein9
The beginning of the 21st century brought a paradigm change in biology. The realization that reductionism has failed to bring about an understanding of complex phenomena has resulted in reappraisals of old research concepts in embryology and cancer research. We will briefly review the historical and theoretical underpinnings of this paradigmatic change because they are central to the study of fetal origins of adult disease in general and the fetal xenoestrogen exposure syndrome in particular.
Origins of evo–devo
In the middle and late 19th century, the main interest of embryologists was the role of the environment in determining a phenotype.Reference Gilbert10 Embryologists frequently observed that nutrition and temperature determined phenotype. From their perspective, the environment was not merely acting on selection of the existing variation (due to different genotypes); rather, the environment was an active determinant of specific adaptations needed to increase fitness. Scott Gilbert identified the development of laboratory animal models as the main circumstance that led embryologists to depart from this tradition in the 20th century. Uniform temperature, invariable light cycles and nutrition ad libitum obliterated the manifestation of environmental influences when animals were raised in the laboratory.Reference Gilbert10 The philosopher and historian Lenny Moss also identified a change of perception in the 20th century that led to the ‘phylogenetic turn’ whereby ‘…the gene and the genetic program became understood to be the principal means by which adapted form is acquired; the theater of adaptation changed from that of individual life histories, that is ontogenies, to that of populations over multiple generations, that is phylogenies’.Reference Moss11 Finally, the discovery of the DNA structure in the 50s led to the perception that biology was reduced to chemistry, and to the notion of a genetic program, in which development was reduced to the automatic execution of a program encoded in the DNA.Reference Soto and Sonnenschein12Footnote a
This previously rigid view of development is rapidly changing, as epidemiological studies reveal the developmental plasticity of the human fetus.Reference Barker and Hanson14 Thus, we now witness the rebirth of ecological developmental biology. Several paths have been identified that could mediate environmental cues into the building of a phenotype, namely, (i) the neuroendocrine route, whereby the nervous system monitors the environment and transfers signals to the endocrine system, (ii) the epigenetic route, whereby environmental agents change the methylation pattern of genes, thereby altering their transcriptional capabilities and (iii) direct modulation of gene expression, particularly by hormonally active agents.Reference Gilbert10 Although there is plenty of evidence that environmental cues affect these paths, the ways they produce changes in phenotype is yet unknown. During the last 50 years, the focus on genetics has mapped the involvement of genes in development by switching them on and off; however, this approach could not explain how form is generated. This realization prompted developmental biologists to return to their tradition, which explained morphogenesis in terms of mechanics and physics.Reference Thompson15–Reference Soto, Sonnenschein and Miquel18
Organicism and reductionism: the role of the cell within the organism
Toward the middle of the 19th century, the cell theory introduced the basic concept that the cell is the unit of life.Reference Sonnenschein and Soto19 However, in multicellular organisms, single cells do not have an existence independent of the whole. Organisms and their cells are ontogenetically linked. From the very start of embryonic life, the levels of biological organization are entangled, meaning that a zygote is both a cell and an organism.Reference Soto, Sonnenschein and Miquel18 The reductionist perspective favors bottom-up causation, meaning that cells ‘make’ the organism by proliferating. In contrast, from a holistic view, the organism ‘makes’ cells by facilitating their division, indicating that causality is a top–down event. Finally, the organicist view considers the embryo as a dynamic open system whereby there are bottom–up, top–down, reciprocal and multiple causalities; meaning that there is no unique, exclusive type of developmental causality.Reference Soto, Sonnenschein and Miquel18, Reference Noble20
The organism imposes global constraints, whereas locally biophysical and biochemical interactions among neighboring cells, tissues and cellular environment determine shape. Differential cell movement and differential cell adhesion are the products of variable physical forces within the developing organism. Morphogens, that is, chemicals created in various areas of a developing organism, form a concentration gradient as they disperse, causing cells that receive dissimilar local concentrations to enter distinct developmental pathways. Such a developing system is not a thing, but a process. It is this dynamic property of the organism that results in level entanglement, as exemplified by the dual nature of the zygote, which is a cell and an organism.Reference Soto, Sonnenschein and Miquel18
How does cancer begin?
In the 19th century, cancer was viewed in the context of the relationship of cells within the organism, and carcinogenesis was viewed in this organicist context. Theories of carcinogenesis then centered on ontogenesis by viewing cancer as a problem of development. For example, Conheim saw cancer as a product of embryonic rests, while Ribbert viewed it as the product of a failure of the restraints exerted by the tissue upon its cells.Reference Triolo21 As recalled above, at the beginning of the 20th century, a change of stance, the ‘phylogenetic turn’, took place and the center of attention focused on the interior of the cell and the dominant view on carcinogenesis was the somatic mutation theory. From this perspective, cancer is a cell-based disease caused by mutations in the DNA of a single founder cell.Reference Hahn and Weinberg22 The research program emanating from this theory has yet to explain how cancer arises and has failed to provide successful therapies. The re-interpretation of evolutionary trends (proliferation as the default state of all cells) has enhanced interest in the earlier focus on the tissue level of organization and the updated tissue organization field theory of carcinogenesis and neoplasia has been gaining momentum.Reference Soto and Sonnenschein23, Reference Baker, Cappuccio and Potter24 A central motif in this theory is the persistence of morphogenic fields throughout adult life; these fields orchestrate histogenesis and organogenesis before birth as well as tissue maintenance and regeneration throughout postnatal life. The tissue organization field theory posits that neoplasms result from a flawed interaction among cells and tissues and that carcinogenesis is potentially reversible.Reference Maffini, Calabro, Soto and Sonnenschein25
The new emphasis on eco–devo and carcinogenesis as a problem akin to development gone awry has prompted scientists to hypothesize that fetal exposure to xenoestrogens may be an underlying cause of the increased incidence of uterine leiomyoma, testicular cancer and breast cancer observed in European and US populations over the past 50 years.Reference Skakkebaek, Meyts and Jorgensen26, Reference Soto and Sonnenschein27
The developmental xenoestrogen exposure syndrome
During the years between 1948 and 1971, the synthetic estrogen diethylstilbestrol (DES) was administered therapeutically to prevent spontaneous abortion.Reference Herbst and Bern6, Reference Mittendorf28 The practice was stopped when rare pathologies like clear cell adenocarcinoma of the vagina and abnormalities in the uterus, oviduct and cervix were diagnosed in young women who had been exposed to DES in utero.Reference Herbst and Bern6, Reference Herbst and Anderson29 Experiments in rodent models following in utero exposure to DES were able to reproduce strikingly similar abnormalities,Reference Herbst and Bern6, Reference McLachlan, Newbold and Bullock7 and also revealed an increased incidence of mammary cancer and an early onset of cessation of estrus cycles. Two decades later, the same outcomes (increased breast cancer incidence and early menopause) were revealed in women gestationally exposed to DES.Reference Hatch, Troisi and Wise30, Reference Palmer, Wise and Hatch31
The DES syndrome played a central role on the conceptualization of environmental endocrine disruptors and was used as a model to guide research on the effect of exposure to xenoestrogens. Developmental exposure to the xenoestrogen bisphenol-A (BPA) in mice caused a complex array of effects that resembled those observed after developmental exposure to DES. Exposure to environmentally relevant doses of BPA during pregnancy alone and during pregnancy through lactation induced both earlier vaginal opening and earlier first estrous and altered estrous cyclicity in offspring. Exposed animals also showed an increase in the number of blood-filled ovarian bursae at 6 months of age, which are thought to be indicative of advanced reproductive aging.Reference Markey, Coombs, Sonnenschein and Soto1 In the mammary gland, it induced preneoplastic lesions in miceReference Vandenberg, Maffini and Schaeberle2 and carcinoma in situ in rats.Reference Murray, Maffini, Ucci, Sonnenschein and Soto3 In addition, when BPA exposure was continued into lactation, it induced increased body weight (BW). Not surprisingly, neonatal exposure to DES also induces obesity.Reference Newbold, Padilla-Banks, Snyder and Jefferson4
Mammary gland development: a lifelong process
Like other ectodermal appendages such as feathers, teeth, hair and salivary glands, the fetal mammary gland is formed by reciprocal interactions between multiple tissue compartments. The mammary epithelium, derived from the embryonic ectoderm, and the mammary mesenchyme, derived from the embryonic mesoderm, are first detected in the mouse between embryonic days (E)10 and E11. From E11 to E12.5, the epithelial placodes increase in size to form rounded buds. Of particular interest is the expression of estrogen receptor α and estrogen receptor β mRNA in the mesenchyme during this period (E12.5–14.5).Reference Lemmen, Broekhof and Kuiper32 Between E13 and E15, the epithelial buds invaginate into the underlying mesenchymal tissue. At the same time, the mesenchyme abutting the mammary epithelium becomes denser than the surrounding mesenchyme with several concentric layers of fibroblasts aligning themselves around the epithelial compartment. At E15.5, the bud elongates to become a cord, the mammary sprout, which invades the underlying fat pad precursor. Branching of the epithelial cord starts at E16.Reference Veltmaat, Mailleux, Thiery and Bellusci33 By E18, branching is apparent and the ductal lumen begins to form.Reference Vandenberg, Maffini and Wadia34 Ductal elongation and branching slow down soon after birth; that is, the gland grows isometrically until puberty.Reference Veltmaat, Mailleux, Thiery and Bellusci33
Estrogens drive massive ductal growth in the peripubertal mammary gland. The ductal ends develop into terminal end buds (TEBs); these are the structures that mediate ductal growth by invasion of the stroma. They become bulbous and show both high proliferative and apoptotic activity. Death of the body cells in the TEBs is essential for the formation of the lumen on the proximal side of the TEBs and for the growth of the subtending duct.Reference Humphreys, Krajewska and Krnacik35 Thus, the ductal tree migrates into the stroma, led by a front of large TEBs. When the ductal tree reaches the edge of the fat pad, it eventually establishes a network of ducts, terminal ducts and a few alveolar buds. Once again, this morphology remains relatively quiescent; minor fluctuations occur with each estrous cycle, adding and removing alveolar buds, until pregnancy. During pregnancy, the entire epithelial compartment undergoes dramatic proliferation resulting in a plethora of alveolar buds and lobulo-alveolar units in preparation for lactation. Once the period of lactation is over, the mammary gland undergoes rapid involution to return to its pre-pregnancy state, a process associated with widespread apoptosis.Reference Daniel and Smith36
BPA and cancer
About 15 years ago, we performed experiments examining the effect of perinatal exposure to BPA on various endocrine organs and the reproductive tract of Sprague-Dawley rats.Reference Rubin, Murray, Damassa, King and Soto5 Mammary tumors frequently develop in aged animals of this strain. We observed more tumors in the BPA-exposed animals than in their controls. This observation prompted us to collect mammary glands in all subsequent experiments both in the rat and mouse models. A significantly different phenotype was revealed by observing the entire mammary gland of adult animals exposed to BPA during fetal development when compared to vehicle-exposed controls. In addition, we examined mammary gland development from the fetal stage through 1.2 years of age. We hypothesized that in adult animals, the florid phenotype observed was due to (a) a direct action of BPA in the mammary gland anlagen, which altered the morphogenesis of the gland, and (b) an indirect effect induced by BPA on the hypothalamic-hypophyseal-ovarian axis that regulates mammotropic hormone secretionReference Soto and Sonnenschein37 (Fig. 1).
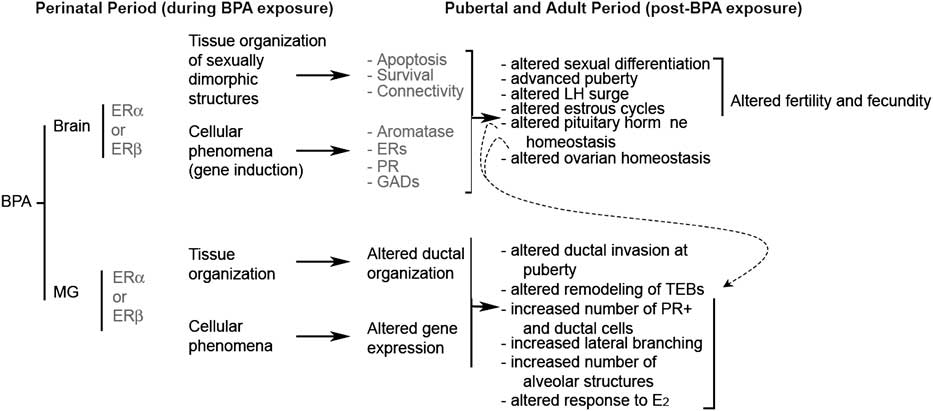
Fig. 1 Effects of bisphenol-A (BPA) exposure during development on female reproduction and mammary gland carcinogenesis. Solid arrows represent pathways whereby BPA affects directly the development of the mammary gland and of sexually dimorphic structures of the brain. Dashed arrows represent secondary effects in the mammary gland mediated by organizational changes in the hypothalamus. The proposed mediators of these outcomes are indicated in red.
The road to cancer: BPA alters the development of the fetal mammary gland
Exposure to BPA from E8 to E18 has significant effects on the mesenchyme, the compartment where estrogen receptors are then expressed. It promotes maturation of the fat pad, an event necessary for ductal invasion and branching. BPA exposure also alters the localization of collagen fibers by increasing the density of collagen fibers directly abutting the epithelium in BPA-exposed mammary glands, while the density of collagen in the entire stromal compartment is significantly decreased in BPA-exposed females when compared with controls. Within the epithelium, BPA exposure leads to decreased cell size, delayed lumen formation, increased ductal area and ductal extensionReference Vandenberg, Maffini and Wadia34 (Fig. 2). Since mammary gland development is dependent on reciprocal interactions between these compartments, the advanced maturation of the fat pad and changes in the extracellular matrix may be responsible for the altered growth, cell size and inhibition of lumen formation observed in the epithelium. In summary, BPA does not uniformly accelerate the morphological process; instead, it disrupts it by advancing some events (i.e. fat pad maturation) while delaying others (i.e. lumen formation).

Fig. 2 Bisphenol-A (BPA) alters the organization of the mammary gland anlagen. Dams were exposed to vehicle or to BPA from gestational day 8 to birth. Upper panel: whole mount of the mammary gland at E18 from a vehicle-exposed female fetus. Lower panel, whole mount of the mammary gland from a female fetus exposed to 250 ng BPA/kg body weight/day. BPA-exposure significantly increased ductal area and ductal extension. Scale bar = 0.5 μm.
Altered morphogenesis continues long beyond the end of the BPA exposure period
In animals exposed to BPA in utero, the discordance between events in the epithelium and stroma persists through postnatal life. For example, at postnatal day 10, the number of epithelial cells undergoing DNA synthesis is significantly reduced in the BPA-exposed animals, while at 30 days of age DNA synthesis in stromal cells is inhibited.Reference Markey, Luque, Munoz de Toro, Sonnenschein and Soto38 At puberty, animals exposed to BPA during fetal development show a decreased rate of ductal invasion of the stroma, probably due to a marked inhibition of apoptosis in the TEB epithelium, a process that results in lumen patency, and thus a decrease in growth of the subtending ducts. Concomitantly, there is an increase in the area and number of TEBs per ductal area, also indicating impaired ductal growth.Reference Munoz de Toro, Markey and Wadia39 There is also an increase in progesterone-receptor positive cells lining the ducts, which are organized in clusters, an indication of presumptive branching points. This is followed by a significant increase in branching at 4 months of age.Reference Munoz de Toro, Markey and Wadia39
The morphological changes found in 30-day-old animals exposed perinatally to BPA could be attributed, at least in part, to an increased sensitivity of the mammary gland to circulating estrogens. Indeed, the magnitude of the response to E2 is significantly enhanced in their siblings that are ovariectomized and exposed to E2 for 10 days.Reference Wadia, Vandenberg and Schaeberle40 These results suggest that increased sensitivity to estrogens drives the induction of progesterone receptor expression in epithelial cells, leading to an increase in lateral branching. By 6 months of age, perinatally exposed virgin mice exhibit mammary glands that resemble those of a pregnant mouse (significant increase in the percentage of ducts, terminal ends, terminal ducts and alveolar buds).Reference Markey, Luque, Munoz de Toro, Sonnenschein and Soto38 These BPA-induced changes in mammary gland development are consistent with the notion that the untimely prenatal exposure to estrogens may predispose the tissue to cancer. Indeed, the persistence of epithelial structures, such as the TEBs and terminal ducts, has been associated with increased carcinogenesis in both rodents and humans.Reference Rothschild, Boylan, Calhoon and Vonderhaar41, Reference Schor, Schor, Howell and Haggie42 Moreover, about 30% of animals that were exposed to similar low doses of BPA during gestation and until postnatal day 16 developed intraductal hyperplasias. The stroma surrounding the ducts bearing intraductal hyperplasias was abnormally collagen-rich.Reference Vandenberg, Maffini and Schaeberle2 This implies the persistence of altered stromal–epithelial interaction beyond the period of exposure, and suggests that understanding the mechanisms of these alterations during fetal life will also shed light on the development of the neoplastic phenotype observed in adulthood.
From pre-neoplastic lesions to cancer
The identification of pre-neoplastic lesions in the mammary glands of mice prenatally exposed to BPA encouraged us to study a rat model because it more closely mimics the human disease with regard to hormone factors and histopathology. Wistar-Furth rats prenatally exposed to 2.5 μg BPA/kg BW/day administered via subcutaneous osmotic pumps to their mothers resulted in a significantly increased number of intraductal hyperplasias (pre-cancerous lesions) observed at 50 days of age and later, while higher doses induced the development of carcinomas in situ (25–33% incidence).Reference Murray, Maffini, Ucci, Sonnenschein and Soto3 In addition, female Wistar rats exposed prenatally to 250 μg BPA/kg BW/day showed an increased proliferation/apoptosis ratio in the epithelial compartment at puberty. During adulthood, those BPA-exposed rats showed an increased number of hyperplastic ducts and the stroma associated with these ducts showed signs of desmoplasia and an increased number of mast cells, suggesting a heightened risk of neoplastic transformation.Reference Durando, Kass and Piva43 In addition, animals prenatally exposed to BPA developed palpable tumors when injected at 50 days of age with the chemical carcinogen nitrosomethylurea while animals not exposed to BPA did not.Reference Durando, Kass and Piva43 In another study, Sprague–Dawley rats were exposed to 250 μg BPA/kg BW/day via daily gavage at post partum days 2 to 20. These perinatally exposed pups showed signs of increased cell proliferation and decreased apoptosis in their mammary gland at puberty. Subsequent exposure to the carcinogen 7, 12, dimethylbenzanthracene at 50 days of age resulted in an increased tumor incidence per animal and a decreased latency period. Invasive cancer was apparent only after administration of a chemical carcinogen at puberty.Reference Jenkins, Raghuraman and Eltoum44 Even though these studies used different rat strains, exposure routes, exposure levels and end points, they all revealed an increased propensity for neoplastic development.
The appearance of these lesions at puberty is reminiscent of the timing of appearance of DES-induced clear cell carcinoma of the vagina in humans, which manifested with a peak incidence at 19 years of age, suggesting that exposure to ovarian hormones contributes to the development of these pathologies. In the mammary gland, the peripubertal period is characterized by intense ductal morphogenesis encompassing tissue remodeling, epithelial invasion of the stroma and increased rates of cell proliferation and cell death, making the pubertal mammary gland particularly prone to neoplastic development. Indeed, the rat mammary gland is especially vulnerable to chemical carcinogenesis during the peripubertal periodReference Russo and Russo45, Reference Grubbs, Peckham and Cato46 and the human mammary gland is known to be especially sensitive to irradiation at this time.Reference Land, Tokunaga and Koyama47
Toward an explanation linking altered development and neoplasia
Supporters of the novel theory of fetal origins of adult disease propose that epigenetic changes such as DNA methylation and chromatin remodeling play a central role in transducing perturbations of the fetal environment into the disease outcomes. For example, permanent alterations in DNA methylation patterns of multiple cell signaling genes identified in the BPA-exposed prostates have been postulated to be the underlying cause of neoplastic development later in life.Reference Ho, Tang, Belmonte de Frausto and Prins48 Like the somatic mutation theory, epigenetic theories of carcinogenesis imply that cancer originates in a single founder cell that undergoes genetic and/or epigenetic changes, which ultimately result in dysregulated cell proliferation.Reference Weinberg49 As mentioned above, the tissue organization field theory postulates, instead, that carcinogenesis represents a problem of tissue organization comparable to organogenesis gone awry.Reference Sonnenschein and Soto19, Reference Sonnenschein and Soto50 Accordingly, carcinogens and teratogens would disrupt normal dynamic interactions of neighboring cells and tissues during early development and/or adulthood.Reference Maffini, Calabro, Soto and Sonnenschein25, Reference Maffini, Soto, Calabro, Ucci and Sonnenschein51 As a result of this disruption in tissue organization, cells would regain their constitutive ability to proliferate and promote neoplastic development.
DES-induced clear cell carcinoma of the vagina also provides an example of tumor development that is likely due to the disruption of tissue organization. This carcinoma originates in areas of cervico–vaginal adenosis, which are regions of simple columnar epithelium that develop within the stratified squamous epithelium of the vagina.Reference Robboy, Welch and Young52, Reference Newbold and McLachlan53 Cervico–vaginal adenosis has been linked to aberrant cell-fate determination whereby vaginal epithelial cells acquire a uterine fate and become a simple columnar epithelium rather than a stratified squamous one.Reference Kurita, Mills and Cunha54 The mesenchyme plays a major role in this epithelial fate determination process.Reference Cunha55 DES may also act directly on the vaginal epithelial cells by blocking the expression of p63, a protein that plays a major role in the fate determination of the vaginal and other stratified squamous epithelia.Reference Kurita, Mills and Cunha54 More recently, msx2, which plays a critical role in cell-fate determination in the vaginal epithelium, was shown to be repressed by DES.Reference Yin, Lin and Ma56 This homeobox-domain transcription factor is required for the correct expression of wnt7a, and the absence of msx2 would result in a complete failure of stratification of the vaginal epithelium.Reference Yin, Lin and Ma56
In contrast with the genital tract, knowledge about how estrogens and other endocrine disruptors affect mammary gland morphogenesis is limited. During the fetal period of exposure to BPA, estrogen receptors are only expressed in the mesenchymal cells. Hence, it is likely that the changes observed in the epithelium are entirely mediated by BPA action on the stroma. Since maturation of the fat pad is the driving event for ductal growth and branching, the increased ductal area in BPA-exposed mice might be due to the acceleration of this process (Fig. 3). Similar to the female genital tract, several members of the wnt signaling cascade and msx2 are expressed during fetal mammary gland development.Reference Veltmaat, Mailleux, Thiery and Bellusci33 The expression of wnt4, wnt5b and msx2 is regulated by estrogens in the adult mammary gland.Reference Weber-Hall, Phippard, Niemeyer and Dale57, Reference Phippard, Weber-Hall and Sharpe58 Hence, it is plausible that fetal xenoestrogen exposure results in the extemporaneous expression of this set of genes that, in turn, may indirectly cause altered morphogenesis and neoplastic development as is the case for clear cell carcinoma of the vagina.

Fig. 3 Postulated causal links connecting perinatal exposure to bisphenol-A (BPA) with the development of pre-neoplastic lesions that manifest in adulthood. The full arrows link observations at embryonic day l8 with postulated causal links based on experimental observations from the literature. Dashed arrows link the observed effects at E18 with effects of prenatal BPA exposure observed during puberty and adulthood.
It is also conceivable that fetal BPA exposure may alter the methylation pattern of genes involved in the reciprocal tissue interactions that mediate morphogenesis. In the prostate, developmental exposure to BPA causes changes in DNA methylation,Reference Ho, Tang, Belmonte de Frausto and Prins48 which also changes with age, suggesting that those changing patterns may instead represent a downstream event in a causal chain. It should be noted that DNA for methylation analysis is usually extracted from the whole tissue, which is morphologically heterogeneous and asymmetric. This may preclude the detection of truly significant changes occurring in structures like the presumptive fat pad, the epithelial leading edge, and the peri-epithelial mesenchyme. Alternatively, some relevant changes in gene expression may occur by means of induction and repression rather than silencing. In addition, important changes may not only be due to altered gene expression, but also to post-transcriptional and/or post-translational regulation as well. For example, breast interlobular and intralobular stroma had similar patterns of mRNA expression while showing significant differences in the expression levels of certain proteins and a very different matrix. In this latter regard, collagen I was more abundant in the interlobular stroma than in the intralobular stroma.Reference Fleming, Long and Ginsburg59 This is a relevant finding since mammographic density is considered to be a strong indicator of breast cancer risk, and this density correlates with stromal density due to collagen deposition.Reference Martin and Boyd60 Moreover, increased stromal collagen density significantly increased tumor formation and metastasis in a mouse transgenic model.Reference Provenzano, Inman and Eliceiri61
Biomechanics and tissue organization
Bioengineers are studying the role of physical forces with the purpose of using these properties to build tissues for transplantation and reconstruction, while developmental biologists are interested in understanding how shapes are determined during histogenesis and organogenesis. Biophysical factors are powerful determinants of tissue organization; this has been known and used therapeutically, for instance, to reshape bones. The advent of tools that measure mechanical forces in soft tissue allows for the study of biomechanics in morphogenesis. Biochemical modulators such as morphogens interact and even transduce into mechanical force, while deformation caused by stretching, compression and shear are transduced into biochemical changes. So far, it is known that increased matrix rigidity results in altered organization of the mammary epithelium, resulting in solid structures that resemble carcinomas,Reference Paszek and Weaver62, Reference Paszek, Zahir and Johnson63 and that matrix rigidity inhibits lumen formation.Reference Paszek, Zahir and Johnson63 Our findings that prenatal exposure of mice to BPA resulted in changes in collagen deposition and organization suggests that matrix rigidity and fiber organization may be one of the early determinants of the altered tissue organization observed during fetal lifeReference Vandenberg, Maffini and Wadia34 (Fig. 3).
Conclusions
Development and carcinogenesis are the result of reciprocal tissue interactions mediated by biophysical and biochemical modulators. These processes involve multiple and complex causality, which cannot be tackled by linear thought and reductionism. Systems biology approaches using mathematical modeling tools and computer simulations may help in the understanding of these complex phenomena. Finally, while research should continue to provide a better understanding of these biological phenomena, it is now clear that the weight of the evidence collected to date favors a swift and effective change in public health and environmental policies aimed at protecting the public at large, and the developing fetus and women of reproductive age, in particular. In this regard, The Endocrine Society Statement succinctly describes what should be done to reach those goals, namely, ‘…to increase understanding of the effects of endocrine disrupting chemicals, including enhancing increased basic and clinical research, invoking the precautionary principle, and advocating involvement of individual and scientific society stakeholders in communicating and implementing changes in public policy and awareness’.Reference Diamanti-Kandarakis, Bourguignon and Giudice64
Acknowledgments
This work was supported by grants from the NIH (ES0150182, ES012301, ES08314 and ES018822) and the Parsemus Foundation. We are grateful to Cheryl Schaeberle for her excellent editorial assistance.
Statement of Interest
None



