INTRODUCTION
Mutations are the raw material of evolution wherein the effect of natural selection depends strongly on the fitness of each mutant within a given environment and genetic background. To understand the nature of quantitative variation, and thus the potential and speed of adaptation of cultivars to different environments (Badu-Apraku et al. Reference Badu-Apraku, Oyekunle, Obeng-Antwi, Osuman, Ado, Coulibay, Yallou, Abdulai, Boakyewaa and Didjeira2012), it is important to determine the positive or negative fitness effects of mutations. Indeed, most mutations affecting fitness and fitness components are harmful (Garcia-Dorado et al. Reference Garcia-Dorado, Monedero and Lopez Fanjul1998). Some mutants reduce fitness, as is the case of the recurrent mutant gene sugary1 (su1) in maize (Zea mays L.) (Revilla et al. Reference Revilla, Malvar, Abuin, Ordás, Soengas and Ordás2000, Reference Revilla, Malvar, Rodriguez, Butrón, Ordás and Ordás2006) located on chromosome 4 (Tracy et al. Reference Tracy, Whitt and Buckler2006). Mutant seeds homozygous for the allele su1 are deficient in the production of insoluble starch, but accumulate an increased proportion of soluble sugars during endosperm development (Schultz & Juvik Reference Schultz and Juvik2004). Sweet corn varieties are cultivars homozygous for su1 (or some other endosperm mutant) and are one of the main products obtained from maize which, in addition to varieties used for cornflakes (Alonso Ferro et al. Reference Alonso Ferro, Malvar, Revilla, Ordas, Castro and Moreno-Gonzalez2008), are directly used for human consumption in temperate areas.
The gene su1 is considered lethal or semi-lethal when introduced in most field maize genetic backgrounds (Tracy Reference Tracy1990). Directional selection against su1 has been reported by Martins & Da Silva (Reference Martins and Da Silva1998) in crosses between Su1 and su1 maize inbred lines. They also found that the reduced germination and smaller seedling vigour of a homozygous su1 seed significantly affect gene frequencies. Revilla et al. (Reference Revilla, Malvar, Abuin, Ordás, Soengas and Ordás2000, Reference Revilla, Malvar, Rodriguez, Butrón, Ordás and Ordás2006, Reference Revilla, Malvar, Ordás, Rodríguez and Ordás2010) and Ordás et al. (Reference Ordás, Rodríguez, Romay, Malvar, Ordás and Revilla2010) also reported that the viability of the su1 and shrunken2 (sh2) mutants depend on specific sweet corn×field maize genotype interaction, with genetic background playing a major role in the viability of those mutants. The same conclusions were reached by Yamamoto et al. (Reference Yamamoto, Anholt and Mackay2009), Le Gac & Doebeli (Reference Le Gac and Doebeli2010) and Magwire et al. (Reference Magwire, Yamamoto, Carbone, Roshina, Symonenko, Pasyukova, Morozova and Mackay2010) for the factors affecting variation of mutant fitness in Drosophila melanogaster. Butler (Reference Butler1977) reported that the mutants whose viability value showed great heterogeneity, with both excesses and deficiencies, were probably influenced by their linkage with other genes. Recently, the genetic effects on the fitness of the su1 allele in wild-type maize were monitored through five successive generations of selfing in two separate designs of mean generation analyses by Djemel et al. (Reference Djemel, Ordás, Khelifi, Ordás and Revilla2011). Fitness of su1 is under genetic control with significant additive effects that are probably due to multiple genes with minor contributions. This suggests that the interaction of genetic backgrounds with alleles could have evolutionary implications by increasing or decreasing the probability of mutant fixation. All the works previously mentioned suggest also that the viability of seeds homozygous for su1 is not solely a function of the allele, but that it is also controlled by other genes.
In order to understand the molecular basis of phenotypic variation in maize, McMullen et al. (Reference McMullen, Kresovich, Villeda, Bradbury, Li, Sun, Flint-Garcia, Thornsberry, Acharya, Bottoms, Brown, Browne, Eller, Guill, Harjes, Kroon, Lepak, Mitchell, Peterson, Pressoir, Romero, Rosas, Salvo, Yates, Hanson, Jones, Smith, Glaubitz, Goodman, Ware, Holland and Buckler2009) crossed 25 diverse inbred lines with the reference inbred line B73 and obtained recombinant inbred lines (RIL) populations to create the Nested Association Mapping (NAM) population; the two sweet corn lines (Il14h and P39) show distortion on chromosome 4 against the su1 mutant. Numerous analyses have demonstrated that context dependency and dynamics variation of mutation across genomes can be attributed to the composition of the nucleotides flanking a mutation (Morton et al. Reference Morton, Bi, McMullen and Gaut2006).
There are several questions that must be answered in order to understand which genes or genetics factors are essential for a mutant viability. Because of the complexity of the multitude of biological processes required for a plant to grow, it is possible that a large and diverse set of genes are likely to be involved. Given the different mechanisms affecting mutant fitness and the need to understand the genetic networks underlying each mechanism, there is a clear need for genome screens to identify genes and genetic networks affecting mutant viability. The aims of the present study were: (i) to determine the size of the critical genomic regions that co-segregates with the su1 allele and examine the existence of chromosomal regions that exhibit segregation distortion against the su1 allele; and (ii) to identify the genes affecting the viability of lines containing the allele su1.
MATERIALS AND METHODS
Single nucleotide polymorphism (SNP) characterization in the RIL
In order to estimate the size of the chromosomal regions flanking the su1 allele, the genetic map of two populations of RILs derived from the cross between the reference inbred B73 (released from the Iowa Stiff Stalk Synthetic population) and two sweet corn inbreds, namely P39 (developed from cvar Golden Bantam) and Il14h (developed from cvar Stowell's Evergreen), was used. Two other RIL populations derived from the cross of B73 with two field maize inbred lines (Oh43 and Tx303) were used as reference. All these materials were part of the NAM population, released and genotyped by the Maize Diversity Project (http://www.panzea.org; now known as Genetic Architecture of Maize and Teosinte; verified 9 March 2012).
The NAM genetic map consists of 1106 loci, with an average marker density of one marker every 1.3 centimorgans (cM) (McMullen et al. Reference McMullen, Kresovich, Villeda, Bradbury, Li, Sun, Flint-Garcia, Thornsberry, Acharya, Bottoms, Brown, Browne, Eller, Guill, Harjes, Kroon, Lepak, Mitchell, Peterson, Pressoir, Romero, Rosas, Salvo, Yates, Hanson, Jones, Smith, Glaubitz, Goodman, Ware, Holland and Buckler2009). As the Su1 locus was not mapped in NAM, the B73 reference genome v2 (http://www.maizesequence.org; verified 9 March 2012) was used to estimate the exact coordinates of this locus and the position on NAM genetic map. The Su1 locus is estimated between the positions 53·7 cM and 55·2 cM on the chromosome 4 and flanked by the markers PZA01751·2 and PZA00445·22. The RILs were classified into field maize or sweet corn types when both flanking markers of Su1 had the B73 or the alternative allele from the sweet corn inbred line (P39 or Il14h), respectively. All intervals with missing values were excluded from the analyses.
Simple sequence repeat (SSR) characterization of the F2 population
To identify the genetic factors controlling the viability of seeds containing different alleles, an F2 population derived from the cross between the field maize inbred line A619 (Lancaster) and the sweet corn inbred line P39 was employed; this population was used because A619 showed the highest negative selection intensity against su1 in a previous study (Djemel et al. Reference Djemel, Ordás, Khelifi, Ordás and Revilla2011).
Self-pollination of the F1 derived from the cross A619×P39 yielded a F2 population in Pontevedra (Spain, 42° 24′ N, 8° 38′ W, 20 m asl) in 2006. Six hundred F2:3 seeds were sown in 2009. The distance was 0·80 m between rows and 0·21 m between plants for a planting density of c. 60000 plants/ha. The F2 plants were self-pollinated. At harvest, the total number of plants that survived was 488, of which 175 were heterozygous Su1su1. All analyses were carried out on heterozygous plants only.
Observations were made on individual plants of: early vigour (at the five-leaf stage by using a visual scale from 1=poor to 9=excellent), leaf chlorophyll content measured at vegetative and reproductive stage using a hand-held Chlorophyll Content Meter, the CCM-200 (Opti-Sciences, Tyngsboro, Massachusetts, USA), ear length (mm), ear weight (g), observed number of field maize and sweet corn (su1su1) seeds and seed type (using a visual scale from 1=dent to 4=flint).
For each plant, 25 seeds from each heterozygote ear for each phenotype (sugary or non sugary) were germinated in Petri dishes in a growing chamber at 25 °C. Each Petri dish had 7 ml of distilled water added before closing with parafilm. Characters related to germination were recorded 7 days later (proportion of sugary or non-sugary seeds with roots and coleoptiles). The coefficient of selection against su1 was calculated as the proportion of germination of sugary seeds relative to that from non-sugary ones.
Leaf tissue was collected from the fourth or fifth leaf for each heterozygous plant. DNA was extracted from leaf tissue according to Liu & Whittier (Reference Liu and Whittier1994). SSR amplifications were performed as described by Butrón et al. (Reference Butrón, Tarrio, Revilla, Malvar and Ordás2003). SSR products were separated after amplification by electrophoresis on a 60 g/l (6%) non-denaturing acrylamide gel (c. 250 V for 3 h) (Wang et al. Reference Wang, Shi, Carlson, Cregan, Ward and Diers2003). A set of 295 SSR markers distributed along the genome were screened for polymorphism among parental inbred lines.
Data analyses
For the RIL populations, the proportion of expected segregation Su1:su1 is 1:1 when there is no selection. The expected number of sweet corn RILs was compared to the observed number using the chi-square goodness of fit test (χ 2) (P<0·05). The maximum and the minimum of the genomic region flanking a Su1 interval were defined as the length of the chromosome fragment (cM) conserving the B73 or the alternative alleles continuously at both sides of the Su1 locus.
Moreover, to examine the existence of chromosomal regions that exhibited segregation distortion against the su1 allele, SNPs variability was classified into two main types (sweet corn and field maize) and, within each type, the number of RILs sharing the B73 alleles was compared with the number of RILs sharing the sweet corn allele in other loci through the genome. The deviation from the expected number was tested with χ 2 at P<0·05 and at P-value with Bonferroni criterion (P<0·05/N; N=number of SNP markers) using a contingency table (Steel et al. Reference Steel, Torrie and Dickey1997). The –log P for the χ 2 value for segregation of B73 v. the two sweet corn parental alleles was calculated.
For each plant of the population (A619×P39) F2, the expected number of sugary seeds was calculated and compared with the observed number of sugary seeds using the χ 2 test at P<0·05. The SSRs associated with su1 viability were identified by using bulk segregant analysis of su1 frequency (Quarrie et al. Reference Quarrie, Lazic-Jancic, Kovacevic, Steed and Pekic1999). A total of 90 SSRs that showed polymorphism between both parental lines (A619 and P39) were used to genotype the heterozygous (Su1su1) plants that exhibited a lower su1 frequency. The SSRs detected to be associated with low su1 frequency were used to genotype all the heterozygous plants from the F2 population in order to quantify the effects of each potential QTL on the fitness of su1. The PROC GLM program (SAS Institute 2008) was used to detect SSRs associated with the su1 allele viability. Each marker locus was analysed for all previously mentioned traits. Since the number of plants within markers was not equal, the comparisons of means of the allelic classes were carried out using least squares means.
RESULTS
Genomics regions with segregation distortion against su1
In both RIL populations developed from crosses between B73 and sweet corn inbreds (B73×P39 and B73×Il14h), a significant segregation distortion was identified for the B73: alternative allele (P39 or Il14h) for the SNPs flanking the Sugary1 locus (Table 1). The segregation was skewed towards the B73 allele and the lowest number of sweet RILs was observed within the RIL released from B73×Il14h. In contrast, the RILs from B73×Oh43 and from B73×Tx303 displayed the expected Mendelian distribution of B73 and the alternative allele with no significant skewness towards the genotype of Oh43 and Tx303, respectively. To better understand how the su1 allele affects the genome, the SNP genotypes at the genomic regions flanking the Su1 locus interval were analysed. No significant differences were noted in the size of the flanking fragments (data not shown) between all four populations.
Table 1. Segregation distortion at the SNPs flanking the sugary1 locus for B73: alternative genotype (P<0·05) in four RILs populations obtained from crosses between the maize inbred line B73 and four diverse lines (DL)
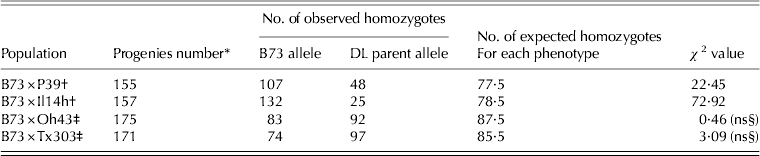
* Excluding RIL with missing value at su1 interval position.
† Sweet corn inbred line.
‡ Field maize inbred line.
§ Not significant at P<0·05.
Using a 0·05 experiment-wise threshold and applying Bonferroni's criterion, only the SNPs located on chromosome 4 showed deviations from the expected frequencies for the B73 or alternate allele in the sugary RIL and non-sugary RIL (Table 2). However, when a 0·05 comparison-wise threshold was applied without Bonferroni's criterion, 0·10 and 0·07 of the markers located outside the chromosome 4 in the B73×P39 and B73×Il14h, respectively, showed that the relative frequency of the B73 or alternate alleles in the sweet corn RIL was different from the relative frequency of those same alleles in the non-sweet maize RIL (Fig. 1). In contrast, when the RILs developed from B73×Oh43 and B73×Tx303 are studied, only 0·01 and 0·05, respectively, of markers located outside the chromosome 4 showed deviations. Depending on the population, the chromosomes showing deviation from the random distribution of B73 and sweet corn alleles were not the same; chromosomes 1, 2, 3, 5, 6, 9 and 10 showed it for B73×P39 and chromosomes 1, 3, 6, 7 and 8 for B73× Il14h. In the absence of both selection and linkage disequilibrium, the segregation of all markers would be expected to follow normal Mendelian frequencies in both sweet and non-sweet RILs. However, all markers were skewed towards the B73 allele in the non sweet corn RILs and towards the P39 or Il14h allele in the sweet corn RILs. In both populations, the maximum distortion was found in chromosome 4. The length of the linkage group of chromosome 4 was larger for Il14h than for P39 (from c. 19·4 to 90·7 cM and from 97·9 to 12·5 cM for Il14h and from 21·3 to 97·9 cM for P39). The SNPs located in chromosomes 1 and 6 were shared by both Il14h and P39, while those in chromosomes 7 and 8 were exclusive to Il14h and those of chromosomes 2, 5, 9 and 10 were exclusive to P39. Only two regions on chromosome 6, i.e. bins 6·07 and 6·08, were common for both populations. All these markers were unevenly distributed over the chromosomes and were located in several regions along the chromosome. The size of these regions varied from c. 1 to 27 cM.

Fig. 1. Segregation distortion across the maize chromosomes (chromosome 4 is not included because the whole chromosome showed segregation distortion). The solid line indicates the –log P for the χ 2 value for segregation of P39 parental allele v. B73 parental allele. The broken line indicates the –log P for the χ 2 value for segregation of Il14h parental allele v. B73 parental allele. The arrow indicates the position of the genomic regions with a significant segregation distortion.
Table 2. SNP markers showing segregation distortion in (B73×P39) and (B73×Il14h) populations for P39* and Il14h* parental alleles v. B73 parental allele, χ 2 value and genetic position (cM) for each marker locus

* Su1 locus is estimated between position 53·7 cM and position 55·2 cM on the NAM map.
† χ 2 value with d.f.=1 and P-value adjusted with the Bonferroni method.
‡ Genetic position in the NAM map (cM).
QTLs related to su1 viability
Bulk segregant analysis was employed in order to detect markers associated with the viability of the su1 mutant along the genome. The χ 2 test detected only eight heterozygous plants that exhibited a lower su1 allele frequency compared with expectations (data not shown). From the 90 SSRs markers used to genotype these plants, only eight were significantly associated with the viability of su1 alleles: phi 090 (Bin 2·08), umc 1746 (Bin 3·01), umc 2259 (Bin 3·03), phi 029 (Bin 3·04), umc 1102 (Bin 3·05), umc 1221 (Bin 5·04), phi 081 (Bin 6·05) and umc 1309 (Bin 8·05) with frequencies that were 0·37, 0·30, 0·31, 0·31, 0·28, 0·31, 0·33 and 0·25 of the allele frequency of P39, respectively. All these SSRs markers were used to genotype all the heterozygous plants from the F2 population to check this association. These analyses revealed significant associations among markers and diverse viability-related traits such as early vigour, ear length, ear weight, number of sugary seeds, seed type and number of seeds with roots and coleoptiles (Table 3). All significant markers explained only low proportions of variability for su1, with R 2 ranging from 0·040 to 0·102. Five SSRs markers were positively associated with number of sugary seeds (umc 1221, R 2=0·048), ear length (phi 081, R 2=0·042), ear weight (phi 029, R 2=0·042), seed type (umc 2259, phi 029 and umc 1221, R 2=0·054, R 2=0·102 and R 2=0·059, respectively), early vigour (phi 029 and umc 1221, R 2=0·040 and R 2=0·045, respectively), proportion of sugary seeds with root (umc 1221, R 2=0·056) and proportion of non-sugary seeds with coleoptiles (umc 1309 and phi 081, R 2=0·079 and R 2=0·065, respectively) (Table 3).
Table 3. Significant main effect of all loci affecting the agronomic traits in the F2 population, P-value, coefficient of determination (R 2) and allelic classes’ effect (means±s.e.)

* Chromosome location.
† Heterozygote class.
‡ Ratio of the su1 endosperm relative to the Su1 endosperm.
Moreover, the loci phi 029, umc 1221 and phi 081 were associated with almost all traits evaluated in the F2 population. The presence of A619 alleles had negative effects on early vigour, number of observed sugary seeds and root growth, while the P39 allele had negative effects on ear length and number of non-sugary seeds with coleoptile (Table 3). The coefficient of selection was calculated as an estimator of su1 fitness; two SSRs markers umc 1221 (R 2=0·057) and phi 081 (R 2=0·051) were associated with this trait. In addition, coefficients of selection were significantly affected by the contribution of a parental allele of A619 (0·71 v. 0·89 and 0·38 v. 1·10 for root growth and coleoptiles growth, respectively).
DISCUSSION
Fitness is not solely a function of the mutant gene but is influenced by other genes. Furthermore, the behaviour of a mutant can vary depending on the context in which the mutation occurs. RILs are formed by crossing two inbred lines followed by repeated selfing to create a random set of inbred lines whose genome is a mosaic of the parental genomes (Yu et al. Reference Yu, Holland, McMullen and Buckler2008). No selection other than natural is applied during the process of developing RILs.
In the two populations of RILs involving sweet corn inbred lines (B73×P39 and B73×Il14h) used in the present study, a net natural selection was revealed acting against the su1 allele, a fact previously reported by McMullen et al. (Reference McMullen, Kresovich, Villeda, Bradbury, Li, Sun, Flint-Garcia, Thornsberry, Acharya, Bottoms, Brown, Browne, Eller, Guill, Harjes, Kroon, Lepak, Mitchell, Peterson, Pressoir, Romero, Rosas, Salvo, Yates, Hanson, Jones, Smith, Glaubitz, Goodman, Ware, Holland and Buckler2009) (Table 1). The sweet corn inbred Il14h brought about a higher reduction of the su1 allele when crossed with B73 than the sweet corn inbred P39 when crossed with the same inbred. The two non-sweet maize inbred lines used as references (Oh43 and Tx303) showed that the effect of selection was solely due to the su1 allele, because the frequency of the alternative allele had a similar frequency as the B73 allele. Furthermore, the number of progenies with a normal segregation for the allele at the Sugary1 locus was lower when the parents were sweet corn lines than when they were not. These results confirm the general observation that the viability of sugary seeds was closely related to the specific sweet×field maize genotype interaction (Revilla et al. Reference Revilla, Malvar, Abuin, Ordás, Soengas and Ordás2000, Reference Revilla, Malvar, Rodriguez, Butrón, Ordás and Ordás2006, Reference Revilla, Malvar, Ordás, Rodríguez and Ordás2010; Djemel et al. Reference Djemel, Ordás, Khelifi, Ordás and Revilla2011).
Fluctuation in the fitness value of genes can be caused by a closely linked gene (Butler Reference Butler1977). In order to understand the genetic regulation of the su1 allele, the variation in the mutation fitness related to the genomic regions flanking the su1 locus was examined. No significant relationship was found between mutation behaviour and size of flanking genome effect; this was probably due to the limited recombination caused by linkage disequilibrium in this region of chromosome 4 (Lu et al. Reference Lu, Romero-Severson and Bernardo2002). Galinat (Reference Galinat and Walden1978) also proposed block inheritance of the genes on chromosome arm 4S, referring to this block as the ‘chromosome 4 complex’, which covers nearly all of 4S from the Ph position to the su1 position. In both populations of the present study, the highest deviation from random distribution was observed in chromosome 4, probably due to the selection against the su1 allele during the selfing generations that yielded the RILs. The Su1 locus was mapped in the chromosome 4 at Bin 4·05 (James et al. Reference James, Robertson and Myers1995). McMullen et al. (Reference McMullen, Kresovich, Villeda, Bradbury, Li, Sun, Flint-Garcia, Thornsberry, Acharya, Bottoms, Brown, Browne, Eller, Guill, Harjes, Kroon, Lepak, Mitchell, Peterson, Pressoir, Romero, Rosas, Salvo, Yates, Hanson, Jones, Smith, Glaubitz, Goodman, Ware, Holland and Buckler2009) and Lu et al. (Reference Lu, Romero-Severson and Bernardo2002) reported that these regions were under higher selective pressure with a low recombination rate, so the Hill–Robertson effect (in a population of finite size which is subject to natural selection, random linkage disequilibria will occur, caused by genetic drift or by mutation, and they will tend to slow down the process of evolution) in a region under strong selection such as this could have increased the linkage block.
In a previous study with maize, a total of 18 chromosomal regions on the 10 maize chromosomes showed segregation distortion (Lu et al. Reference Lu, Romero-Severson and Bernardo2002). In the present study, various SNP markers located outside the chromosome 4 in the two sweet corn RILs populations showed non-random distribution of the allelic frequency of the B73 or alternate alleles in both the sweet corn and the non-sweet maize RILs. These markers were skewed towards the B73 allele in the non-sweet maize RILs and towards the P39 or Il14h allele in the sweet corn RILs. In the study of Eichten et al. (Reference Eichten, Foerster, De Leon, Kai, Yeh, Liu, Jeddeloh, Schnable, Kaeppler and Springer2011), two inbred lines parents (B73 and Mo17) were used to produce a set of near-isogenic lines (NILs). The 150 NILs produced were separated into two background: 100 lines with B73 as the recurrent parent and Mo17 as the donor parent (B73-like NILs), and 50 lines with Mo17 as the recurrent parent and B73 as the donor parent (Mo17-like NILs). Eichten et al. (Reference Eichten, Foerster, De Leon, Kai, Yeh, Liu, Jeddeloh, Schnable, Kaeppler and Springer2011) identified several regions with the opposing biases towards either the B73 or Mo17 parental allele in both background. The most likely explanation is that there are several loci for which there is a preferred allele with the ability to confer increased fitness. This result supports the present authors' suggestion that the alleles from the sweet corn parent are required for guarantying the viability of the su1 allele. All these SNPs were detected in the 10 chromosomes of maize, but they were unequally distributed depending of the RIL population (Fig. 1). The present results indicate that genetic factors for su1 fitness exist on most chromosomes and also that the fitness of an allele depends on the specific genetic background (Revilla et al. Reference Revilla, Malvar, Rodriguez, Butrón, Ordás and Ordás2006). Although the effects of a few genes of large effect on su1 fitness could still be a reasonable hypothesis (Djemel et al. Reference Djemel, Ordás, Khelifi, Ordás and Revilla2011), the results of the present paper suggest that there are probably also a lot of genes with minor effects affecting the diverse viability-related traits.
In order to check this hypothesis, bulk segregant analyses were used to detect markers associated with the su1 mutant fitness along the 10 maize chromosomes in (A619×P39)F2 by using SSR markers. The selection against the mutant may operate either through viability at germination or at the seedling growth stage (Falconer Reference Falconer1981; Martins & Da Silva Reference Martins and Da Silva1998; Ordás et al. Reference Ordás, Rodríguez, Romay, Malvar, Ordás and Revilla2010) and then through reduced fertility (pollen and ovule production, pollination pattern and zygote development) (Clegg et al. Reference Clegg, Kahler and Allard1978; Falconer Reference Falconer1981; Cisneros-López et al. Reference Cisneros-López, Mendoza-Onofre, Zavaleta-Mancera, Gonzalez-Hernandez, Mora-Aguilera, Cordova-Tellez and Hernandez-Martinez2010; Zhang et al. Reference Zhang, Li and Lian2011).
In the present study, the SSR markers detected to be associated with su1 fitness were chosen based on the low frequency of the P39 allele in the bulk segregant analysis. Only heterozygous plants (Su1su1) were used and the su1 allele frequency was calculated after pollination. For this reason, the selection against the su1 allele was probably due only to fertility factors. Significant genomic regions associated with su1 fitness were detected (Table 3) and those markers were tested for the other viability traits. The SSR marker umc 1221 (with R 2=0·048) located on chromosome 5 was strongly associated with the observed number of sugary seeds. This marker was also associated with other important traits: early vigour and proportion of sugary seeds with roots. Interestingly, the effect of the P39 allele was positive for all these traits. This region of the chromosome 5 (Bin 5·04) was also detected in the RIL population B73×P39 and exhibited a net deviation from the random distribution of the P39 allele. The centromeric region of chromosome 5, as well as the other regions (Bin 6·07 and Bin 6·08) with significant deviations from the random distribution detected in both RILs’ populations, have all been reported as regions with major effects on grain yield (Graham et al. Reference Graham, Wolff and Stubert1997; Schaeffer et al. Reference Schaeffer, Byrne and Coe2006; McMullen et al. Reference McMullen, Kresovich, Villeda, Bradbury, Li, Sun, Flint-Garcia, Thornsberry, Acharya, Bottoms, Brown, Browne, Eller, Guill, Harjes, Kroon, Lepak, Mitchell, Peterson, Pressoir, Romero, Rosas, Salvo, Yates, Hanson, Jones, Smith, Glaubitz, Goodman, Ware, Holland and Buckler2009; Schön et al. Reference Schön, Dhillon, Utz and Melchinger2010), so these regions seem to be good candidates to contain genes involved in the regulation of su1 fitness. Further research should be made to deepen knowledge on this matter.
Some other markers were significantly associated with several traits simultaneously. For example, marker phi 029 was associated with early vigour, ear weight and seed type, and marker phi 081 significantly affected ear length and coleoptile development. However, some markers were only associated with one trait, for example umc 2259, that was only related to seed type, and umc 1309 to non-sugary, related to seeds with coleoptile. No marker located on chromosome 4 was significantly associated with the low fitness of su1, probably due to the high linkage disequilibrium in the region neighbour to the su1 locus.
An important consideration is the presence of genomic regions that control both viability and fertility which is a clear evidence of pleiotropic effect on mutant fitness and the close relationship between the two factors of natural selection. Various authors studied the genetic control of germination and seedling as two factors limiting sweet corn cultivation (Gad & Juvik Reference Gad and Juvik2002; Juvik et al. Reference Juvik, Yousef, Han, Tadmor, Azanza, Tracy, Barzur and Rocheford2003). Tracy (Reference Tracy and Hallauer2001) also reported that these two characters are affected by genetic factors, both at planting and during seed production. A powerful indication is the importance of the genetic background effect on the lethality or near-lethality of the su1 allele when it is introduced into field maize. It is proposed that the pleiotropic effects of genes selected for their desired traits by humans probably have a role in this loss of fitness (Keightley & Hill Reference Keightley and Hill1990). The choice of field maize and sweet corn parents affects the relative positive or negative effects of the alleles; indeed, A619 is not a vigorous inbred but has a better agronomic performance than P39; therefore, the allele from A619 had a negative effect on early vigour and coleoptile development, and a positive effect on ear weight and ear length. Furthermore, as was also observed for the P39 and Il14h RILs, each allele performed better in his original genetic background, i.e. the coleoptiles were more abundant when the sugary seeds had the P39 allele and when the non-sugary seeds had the A619 allele (Table 3). The potential QTLs with highest effect on any trait was phi 029 for seed type (R 2=0·102), but most QTLs had R 2∼0·05. The work of Rebourg et al. (Reference Rebourg, Chastanet, Gouesnard, Welcker, Dubreuil and Charcosset2003), based on both molecular and historical data, revealed that the European maize was related to the Northern Flint material, the progenitor of the modern sweet corn (Revilla & Tracy Reference Revilla and Tracy1995). Malvar et al. (Reference Malvar, Revilla, Cartea and Ordás1997) reported that European flint inbreds offer new possibilities for improving the adaptation of sweet corn to European conditions. All of these results suggest that the SSRs associated with seed type can be candidates for su1 adaptation.
The genomic regions identified in the two RILs populations that showed segregation distortion against the su1 allele can be potential candidates for QTLs of mutant fitness. However, the number of SSR markers detected to be associated with su1 fitness in the F2 population was low. The present study employed different marker types. Significant associations between markers and traits were detected with the SSRs, while linkage disequilibrium was detected with the SNPs. The combination of these two marker types in future works can enhance significantly the power to detect QTLs. The genetic regulation of mutant fitness is still poorly understood and further research should be carried out using larger samples and more markers.
It can be concluded that su1 fitness depends on many genes with small effects on a variety of viability-related traits throughout the genome that are significant or not depending on the genetic background of the materials involved.
This research was supported by the Spanish Plan I+D (AGL2007-64218/AGR and AGL2010-22254) and the Excma. Diputación Provincial de Pontevedra. A. Djemel acknowledges his fellowship from the Spanish Council for Scientific Research (CSIC). We thank the members of the E. Buckler laboratory, for providing the genotypic data of NAM population.