



Management Implications
Reynoutria (knotweeds) are targeted for herbicide treatment and mechanical control, and researchers are actively exploring biocontrol solutions to their rapid spread in Europe and North America. In published studies, Reynoutria japonica (syn. Polygonum cuspidatum Siebold & Zucc.; Japanese knotweed) has received the most attention, but Reynoutria invasions also include Reynoutria sachalinensis (syn. Polygonum sachalinense F. Schmidt ex Maxim.; giant knotweed) and a variety of interspecific hybrids. Management approaches can achieve differential success depending on the identity and genetic makeup of the species being targeted. It will be important to evaluate the effectiveness of treatment approaches at least by species, preferably taking into account the genetic makeup of hybrids, where applicable. Our study confirms that Reynoutria species and hybrids are genetically diverse in North America, and we offer a novel approach for identifying them in a meaningful way. We also demonstrate that morphological characters may be less useful in places where hybrid Reynoutria are diverse. Land managers who are interested in evaluating the effectiveness of their control strategies against the genetic identities of noxious Reynoutria plants may benefit from having their target material sequenced for the plastid matK gene and the nuclear LEAFY second intron. Our study was conducted on plants in Wisconsin, but one could reasonably use the same approach to identify Reynoutria plants across the native and introduced ranges.
Introduction
Knotweeds in the genus Reynoutria, native to eastern Asia, are noxious weeds in Europe and North America, where they continue to spread rapidly (Bailey Reference Bailey, Child, Brock, Prach, Pysek, Wade and Williamson2003; Bailey and Wisskirchen Reference Bailey and Wisskirchen2006; Bailey et al. Reference Bailey, Bímová and Mandák2009; Freeman and Hinds Reference Freeman and Hinds2005; Gaskin et al. Reference Gaskin, Schwarzländer, Grevstad, Haverhals, Bourchier and Miller2014; Grimsby et al. Reference Grimsby, Tsirelson, Gammon and Kesseli2007). Invasive Reynoutria plants are notorious for impacting native vegetation through allelopathy (Murrell et al. Reference Murrell, Gerber, Krebs, Parepa, Schaffner and Bossdorf2011; Parepa et al. Reference Parepa, Schaffner and Bossdorf2012; Vrchotová and Šerá Reference Vrchotová and Šerá2008) and for propagating readily through both sexual and asexual means (Bailey et al. Reference Bailey, Bímová and Mandák2009; Brock et al. Reference Brock, Child, de Waal, Wade, Pyšek, Prach, Rejmánek and Wade1995; Forman and Kesseli Reference Forman and Kesseli2003; Hollingsworth and Bailey Reference Hollingsworth and Bailey2000; Pyšek et al. Reference Pyšek, Brock, Bímová, Mandák, Jarošík, Koukolíková, Pergl and Štěpánek2003). Molecular evidence strongly supports the phylogenetic independence of Reynoutria from related genera (Galasso et al. Reference Galasso, Banfi, De Mattia, Grassi, Sgorbati and Labra2009; Sanchez et al. Reference Sanchez, Schuster and Kron2009; Schuster et al. Reference Schuster, Wilson and Kron2011), thus we use nomenclature that regards Reynoutria as a distinct genus in the Polygonaceae. There are only five Reynoutria taxa, most with native geographic ranges that are rather narrow (Bailey Reference Bailey, Child, Brock, Prach, Pysek, Wade and Williamson2003; Steward Reference Steward1930). Three taxa have established as invasive plants in Europe and North America: Japanese knotweed (Reynoutria japonica Houtt.; syn. Polygonum cuspidatum Siebold & Zucc.) from China, Japan, Korea, and Russia; giant knotweed [Reynoutria sachalinensis (F. Schmidt) Nakai; syn. Polygonum sachalinense F. Schmidt ex Maxim.], native to Japan, Korea, and Sakhalin Island (Russia); and Bohemian knotweed (R. × bohemica Chrtek & Chrtková; syn. Polygonum × bohemicum (Chrtek & Chrtková) Zika and Jacobson). The last is a hybrid between R. japonica and R. sachalinensis, comprising natural hybrids in Japan and invasive populations in Europe and North America (Bailey Reference Bailey, Child, Brock, Prach, Pysek, Wade and Williamson2003; Mandák et al. Reference Mandák, Pyšek, Lysák, Suda, Krahulcová and Bímová2003). The invasive Reynoutria taxa are monophyletic and distinct from the other two species in the genus: Reynoutria ciliinervis (Nakai) Moldenke [syn. Polygonum ciliinerve (Nakai) Ohwi] and Reynoutria multiflora (Thunb.) Moldenke (syn. Polygonum multiflorum Thunb.) (Park et al. Reference Park, Bhandari, Won, Park and Park2018).
Both R. japonica and R. sachalinensis are widespread in North America, as are F1 hybrids (i.e., R. × bohemica) and later-generation hybrids or introgressed individuals (Bailey Reference Bailey2013; Forman and Kesseli Reference Forman and Kesseli2003; Gaskin et al. Reference Gaskin, Schwarzländer, Grevstad, Haverhals, Bourchier and Miller2014; Grimsby et al. Reference Grimsby, Tsirelson, Gammon and Kesseli2007). Although the invasive taxa are closely related and have similar ecologies and morphologies, they may respond differently to management applications (e.g., Bímová et al. Reference Bímová, Mandák and Kašparová2004; Clements et al. Reference Clements, Larsen and Grenz2016; Fung et al. Reference Fung, González-Moreno, Pratt, Oliver, Bourchier and González-Suárez2020). Additionally, hybridization and introgression may produce novel genetic combinations that present further management challenges (Bailey et al. Reference Bailey, Bímová and Mandák2007; Clements et al. Reference Clements, Larsen and Grenz2016; Mitchell et al. Reference Mitchell, Owens, Hovick, Rieseberg and Whitney2019; Parepa et al. Reference Parepa, Fischer, Krebs and Bossdorf2014). To implement effective management plans, it will be important to identify species and hybrids correctly. However, it can be difficult to know in the field whether individuals are hybrids or pure parental species because of their morphological similarity and ability to interbreed. Molecular tools can offer a valuable and independent source of characters for identification, and several strategies have been developed. Sequencing or restriction fragment analyses of plastid genes can produce data reliably; however, the uniparentally inherited plastid data can offer evidence of only one parental lineage per plant. A more thorough summary of genetic variation and hybrid parentage can be obtained using data from nuclear gene regions, which are biparentally inherited, may exist as allelic variants, and may be used to reconstruct polyploidization events (Small et al. Reference Small, Cronn and Wendel2004).
Phylogenetic studies of Reynoutria in the introduced range are relatively sparse, and many have relied upon plastid sequence data that cannot distinguish parental species from hybrids. In light of the fact that two species and their hybrid coexist throughout most of the introduced range, it is important to identify taxonomic entities correctly. Nuclear DNA sequence data are surprisingly rare for Reynoutria species and comprise few sequences of the internal transcribed spacer (ITS) region and LEAFY second intron, compared with the relative abundance of plastid sequence data (Gammon and Kesseli Reference Gammon and Kesseli2010; Grimsby and Kesseli Reference Grimsby and Kesseli2010; Hollingsworth et al. Reference Hollingsworth, Bailey, Hollingsworth and Ferris1999). Park et al. (Reference Park, Bhandari, Won, Park and Park2018) conducted an extensive study of genetic variation in native Reynoutria populations over much of Japan and Korea, as well as Sakhalin Island and mainland Russia, and their study also included introduced plants in the United Kingdom and United States. Combined plastid and nuclear data largely supported the reciprocal monophyly of R. japonica and R. sachalinensis, with the exception of plants collected from Korean islands, which resolved among R. sachalinensis on the LEAFY tree but within the clade of R. japonica on the plastid tree (Park et al. Reference Park, Bhandari, Won, Park and Park2018). Additionally, a small number of samples were determined to have polymorphic LEAFY sequences, some reflecting a combination of R. japonica and R. sachalinensis genetic elements. Molecular data also supported the synonymy of Fallopia forbesii (Hance) Yonek. & H. Ohashi with R. japonica.
Hybridization and introgression are well documented among invasive Reynoutria taxa, and in North America there is a rich diversity of genotypes and morphological variation (Gaskin et al. Reference Gaskin, Schwarzländer, Grevstad, Haverhals, Bourchier and Miller2014; Grimsby and Kesseli Reference Grimsby and Kesseli2010; Grimsby et al. Reference Grimsby, Tsirelson, Gammon and Kesseli2007). Nuclear genetic variation has been examined using a variety of methods that analyze sequence length polymorphisms (Bzdega et al. Reference Bzdega, Janiak, Książczyk, Lewandowska, Gancarek, Sliwinska and Tokarska-Guzik2016; Gammon et al. Reference Gammon, Grimsby, Tsirelson and Kesseli2007; Gaskin et al. Reference Gaskin, Schwarzländer, Grevstad, Haverhals, Bourchier and Miller2014; Grimsby and Kesseli Reference Grimsby and Kesseli2010; Grimsby et al. Reference Grimsby, Tsirelson, Gammon and Kesseli2007; Hollingsworth et al. Reference Hollingsworth, Hollingsworth, Jenkins, Bailey and Ferris1998), but for many applications these techniques may be prohibitively expensive or laborious. Moreover, the clustering and principal components analyses that are used to summarize sequence length polymorphism data are not phylogenetic and cannot reconstruct genetic inheritance with the same level of confidence as a phylogenetic analysis of nucleotide sequence data.
North American Reynoutria species are overdue for a phylogenetic analysis that combines plastid and nuclear sequence data and potentially reconstructs the parentage of hybrids. To this end, we investigated Reynoutria plants in Wisconsin, USA, using plastid matK and nuclear LEAFY sequence data to ascertain whether they represent parental species or hybrids and to infer the most closely related plants among those that have been sampled in eastern Asia. Reynoutria plants introduced to the East Coast continue to expand their range westward, and the surge of invasive Reynoutria in Wisconsin is relatively recent (Barney Reference Barney2006; EDDMapS 2020). Both R. japonica and R. sachalinensis are reported from Wisconsin (Figure 1), as well as R. × bohemica and numerous plants that are difficult to identify, possibly as a result of mixed parentage (i.e., hybridization or introgression). The prospect of identifying Reynoutria species using a small number of nucleotide sequences holds the potential to make genetic identification more accessible to land managers (i.e., by decreasing cost and technical difficulty) and to increase the number of plants that are identified in this way, thus improving our understanding of the ecology and geographic distribution of these troublesome plants.

Figure 1. Sampling localities for Reynoutria in Wisconsin. Locality numbers are referenced in Table 1. Underlying shapes depict reported localities for Reynoutria species (EDDMapS 2020). Pie charts show genetic contributions from the plastid matK gene (top half of pie) and the nuclear LEAFY gene (bottom half of pie); plants with different LEAFY sequence types have different colors in the bottom half.
Materials and Methods
Reynoutria samples were collected from 37 localities across 14 Wisconsin counties (Table 1; Figure 1). Voucher specimens were deposited in the University of Wisconsin–Whitewater herbarium (UWW). Plants were identified initially using two morphological characters that were previously reported to be diagnostic (Bailey et al. Reference Bailey, Bímová and Mandák2009; Zika and Jacobson Reference Zika and Jacobson2003). Leaf laminar bases were scored as truncate (R. japonica), with lobes >2 cm long (R. sachalinensis), or with lobes <2 cm long (R. × bohemica). Hairs on the abaxial leaf veins were scored as multicellular (R. sachalinensis), stout and unicellular (R. × bohemica), or reduced to rounded bumps, that is, scabrous (R. japonica). Secondary morphological characters included the laminar apex (scored as acuminate or acute) and inflorescence length relative to leaf length.
Table 1. Collection localities and morphological features of Reynoutria specimens used in this study.

We also recorded the reproductive condition of plants, where possible. Authors variously have described Reynoutria plants as gynodioecious (Bailey Reference Bailey, de Waal, Child, Wade and Brock1994; Barney et al. Reference Barney, Tharayil, DiTommaso and Bhowmik2006; Beerling et al. Reference Beerling, Bailey and Conolly1994; Freeman and Hinds Reference Freeman and Hinds2005; Hollingsworth et al. Reference Hollingsworth, Hollingsworth, Jenkins, Bailey and Ferris1998), subdioecious (Forman and Kesseli Reference Forman and Kesseli2003; Grimsby et al. Reference Grimsby, Tsirelson, Gammon and Kesseli2007), functionally dioecious (Kim and Park Reference Kim and Park2000), or dioecious (Decraene and Akeroyd Reference Decraene and Akeroyd1988; Li and Park Reference Li, Park, Wu, Raven and Hong2003). Although the technical definition of the reproductive system in Reynoutria may not be universally agreed upon (or consistent across native and adventive ranges), there is general consensus that individuals are either primarily staminate or fully pistillate. We recorded the reproductive system of collected plants as “male-fertile” if we observed stamens on flowers and as “male-sterile” if no stamens could be located (Bailey et al. Reference Bailey, Child and Conolly1996). By using these categories, we acknowledge that male-fertile plants may have functional gynoecia but potentially at low frequencies that escape detection. Male-fertile plants also were characterized by extensive post-anthetic flower abscission, although it should be noted that flower abscission also has been implicated in seed dispersal on male-sterile plants (Beerling et al. Reference Beerling, Bailey and Conolly1994).
Genomic DNA was extracted from dried or fresh leaf material using a standard CTAB protocol (Doyle and Doyle Reference Doyle and Doyle1987), modified as described previously (Tippery et al. Reference Tippery, Pesch, Murphy and Bautzmann2020). We used primers that other researchers had designed for the plastid matK protein-coding region (AF and 8R; Yan et al. Reference Yan, Pang, Jiao, Zhao, Shen and Zhao2008) and the second intron of the nuclear LEAFY gene (MLFYI2-1543F and MLFYI2-2385R; Schuster et al. Reference Schuster, Wilson and Kron2011). We elected to target the matK and LEAFY genes so that our data could be evaluated in a phylogenetic context against the extensive data that were reported for plants in the native range (Park et al. Reference Park, Bhandari, Won, Park and Park2018). After recovering polymorphic LEAFY sequences for several accessions, we designed two forward primers that would differentially amplify the two kinds of sequence: Japo1F (5′-GGTAAAGTGAGAGACGTATAAATG-3′) and Sach1F (5′-AGCTAGTTAGCTAGGTAGCTAC-3′), named for the similarity of amplicon sequences to reported sequences of R. japonica and R. sachalinensis, respectively (Park et al. Reference Park, Bhandari, Won, Park and Park2018).
The polymerase chain reaction (PCR) was conducted in 10-µl reactions each containing 2 µl of 5× reaction buffer, 0.8 µl of 2.5 mM dNTPs, 0.5 µl of each 10 µM primer, and 0.2 µl of Phire™ Hot Start II DNA Polymerase (Thermo Fisher, Waltham, MA, USA). The thermal cycler protocol consisted of an initial denaturation at 98 C for 30 s, followed by 35 cycles of 90 C for 5 s, 52 C (matK primers) or 58 C (LEAFY primers) for 5 s, and 72 C for 20 s, ending with a final extension at 72 C for 60 s. Sanger sequencing was conducted remotely (Eurofins Genomics, Louisville, KY, USA) and employed the same primers used for PCR.
Sequence files were curated in 4Peaks v. 1.8 (Griekspoor and Groothuis 2006) and exported as text files. Any samples that produced polymorphic LEAFY sequences with the newly designed primers were subcloned into bacteria using the pGEM®-T Easy Vector System (Promega Corporation, Madison, WI, USA), then subjected to PCR and sequencing as described earlier. Outgroup sequences were obtained from GenBank; sequences of R. multiflora (EU024768, HM357918, KJ863098, KJ863100) were used to root the matK tree (Park et al. Reference Park, Bhandari, Won, Park and Park2018; Yu et al. Reference Yu, Fa, Xu, Zhu, Hou, Lin and Li2008), and the LEAFY tree was rooted with sequences from several Muehlenbeckia species (JF831223–JF831230, JF831348–JF831355; Schuster et al. Reference Schuster, Wilson and Kron2011). Newly generated and outgroup sequences were combined with Reynoutria sequences obtained from GenBank and previously published Reynoutria sequences that are not yet deposited in GenBank (Park et al. Reference Park, Bhandari, Won, Park and Park2018), then all sequences for each gene region were aligned manually in Mesquite v. 3.61 (Maddison and Maddison Reference Maddison and Maddison2019). Insertions and deletions (indels) were encoded using simple indel coding (Simmons and Ochoterena Reference Simmons and Ochoterena2000), implemented in the program SeqState v. 1.4.1 (Müller Reference Müller2005).
Separate phylogenetic analyses were run for the plastid and nuclear data matrices, using maximum-likelihood criteria in IQ-TREE v. 2.0.5 (Nguyen et al. Reference Nguyen, Schmidt, von Haeseler and Minh2015) with separate partitions (Chernomor et al. Reference Chernomor, von Haeseler and Minh2016) for nucleotide and binary indel data. ModelFinder (Kalyaanamoorthy et al. Reference Kalyaanamoorthy, Minh, Wong, von Haeseler and Jermiin2017) selected the general time reversible (GTR) model for LEAFY and the transversion model (TVM) for matK, both with empirical frequencies and two rate categories. The indel data were analyzed using the GTR model for binary data with empirical frequencies, two rate categories, and ascertainment bias correction (Lewis Reference Lewis2001). Nodal support values for the maximum-likelihood analysis were obtained by running 1,000 bootstrap replicates. Bayesian phylogenetic analyses were conducted in MrBayes v. 3.2.7 (Ronquist et al. Reference Ronquist, Teslenko, van der Mark, Ayres, Darling, Höhna, Larget, Liu, Suchard and Huelsenbeck2012) using the same models as for nucleotide data and the “standard” model for indel data. Nodal support values were calculated by summarizing the posterior distribution after running 2 million generations and discarding the first 25% of generations as burn-in.
Results and Discussion
On the basis of morphological characters, only one accession (R37) was unambiguously identified as R. sachalinensis, because it had leaves >20 cm long with distinct basal laminar lobes and multicellular abaxial vein hairs (Table 1). In all other accessions, the vein hairs were either stout and unicellular or lacking, as have been described for R. × bohemica and R. japonica, respectively (Zika and Jacobson Reference Zika and Jacobson2003). Laminar apices were almost all acuminate, and laminar bases were predominantly truncate. On the basis of these characters, 16 specimens would be identified as R. japonica (i.e., with truncate laminar bases and scabrous abaxial veins), and 2 specimens (R05, R23) would be identified as R. × bohemica (i.e., with shallowly cordate laminar bases and stout, unicellular hairs). Another 18 specimens exhibited combinations of laminar base and abaxial vein morphologies that were inconsistent with published descriptions (Zika and Jacobson Reference Zika and Jacobson2003). Flowering material was observed on most specimens, the majority of which were scored as male-fertile. Inflorescence length was consistently shorter than leaf length for all flowering specimens, although the available material was collected from apical stem regions, and these regions may not have the same leaf and inflorescence morphologies as midstem regions (Barney et al. Reference Barney, Tharayil, DiTommaso and Bhowmik2006).
Plastid (matK) sequence data were obtained for 34 accessions, and nuclear (LEAFY) sequence data for 35 accessions. Newly generated sequences were deposited in GenBank under accession numbers MW770182–MW770215 (matK) and MW771206–MW771273 (LEAFY) (Table 1). PCR amplifications that used specific LEAFY primers produced products for 32 accessions with the Sach1F primer, and for 29 accessions with the Japo1F primer. Three accessions (R09, R12, R13) produced PCR products only for the Japo1F primer, and six accessions (R04, R16, R24, R35, R36, R37) produced products only for the Sach1F primer. The matK data matrix comprised 78 sequences and 1,602 nucleotide characters, with 68 (4.2%) of these being variable among accessions, and one indel character. The LEAFY data matrix included 126 sequences and 1,667 characters (256, or 15.4% variable), and 96 indels were scored. The phylogeny for matK (Figure 2) clearly differentiated two haplotypes for the specimens in our study. Most plants had the “japonica-type” matK sequence, which had been reported previously for plants in Canada and the United States, as well as native R. japonica plants in Japan and Russia. Plants with the “sachalinensis-type” matK sequence were similar to native R. sachalinensis plants from Japan and one accession from the adventive range in the United Kingdom (EF438009).

Figure 2. Phylogeny of Reynoutria species constructed using data from the plastid matK gene. Previously published sequences are shown with GenBank accession numbers and sample identifiers (Park et al. Reference Park, Bhandari, Won, Park and Park2018). Newly generated sequences are shown in bold and referenced in Table 1. Nodal support values represent maximum-likelihood bootstrap percentage.
The LEAFY phylogeny (Figure 3) resolved six clades of sequence types in Wisconsin plants. Four of these, identified as J1 through J4, resolved within a larger clade of sequences from R. japonica (i.e., “japonica-type”), and two sequence types (S1 and S2) were more closely related to R. sachalinensis (i.e., “sachalinensis-type”) (Park et al. Reference Park, Bhandari, Won, Park and Park2018). Sequence types J1 and J2 were most similar to two sequences cloned from an R. japonica plant collected in Vladivostok, Russia (j_rs01). The relationships of sequence types J3 and J4 to native plants were less certain, and these sequences resolved among plants from Japan and Korea on the LEAFY phylogeny. Three accessions collected in northwestern Wisconsin (R09, R12, R13) were polymorphic for “japonica-type” LEAFY sequences, having a combination of J2/J3/J4 sequence types (Figure 1). Two other accessions also had three types of LEAFY sequence: R25 had the S1/J2/J3 combination, and R31 combined S1/J1/J3. No plants in our study were monomorphic for “japonica-type” leafy sequences, and no individuals had the J1 and J2 sequence types in the same plant.
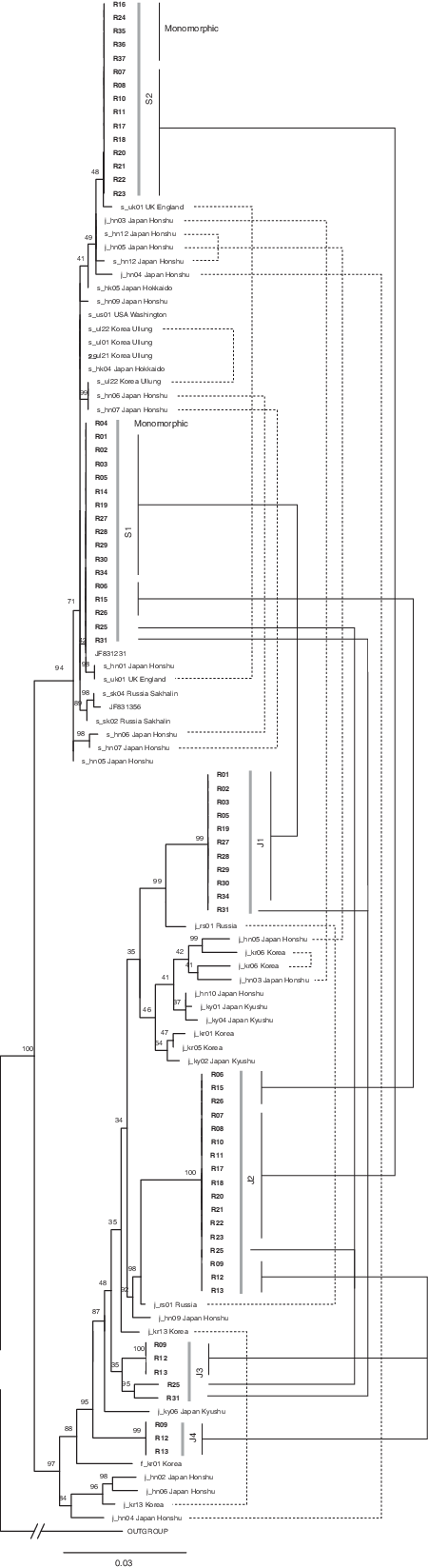
Figure 3. Phylogeny of Reynoutria species constructed using data from the nuclear LEAFY gene. Previously published sequences are shown with GenBank accession numbers or sample identifiers (Park et al. Reference Park, Bhandari, Won, Park and Park2018). Newly generated sequences are shown in bold and referenced in Table 1. Nodal support values represent maximum-likelihood bootstrap percentage. Multiple sequences that were obtained from the same accession are connected by lines at right; dotted lines connect previously reported sequences, and solid lines connect newly reported sequences.
Sequences falling into the R. sachalinensis clade were identified as S1 and S2, and no single individual from Wisconsin was found to have both kinds of “sachalinensis-type” LEAFY sequence. However, five accessions (R16, R24, R35, R36, R37) were monomorphic for the S2 sequence, including two accessions (R24, R37) that also had “sachalinensis-type” matK sequences. Interestingly, in a prior study, one R. sachalinensis plant from the adventive range in Leicester, UK (s_uk01), was found to contain both the S1 and S2 sequence types (Park et al. Reference Park, Bhandari, Won, Park and Park2018). Among plants in the native range, the sequences most similar to S1 and S2 appear in plants from Japan. An invasive R. sachalinensis plant sequenced from Washington, USA (s_us01), was similar but not identical to the S1 sequence type (Park et al. Reference Park, Bhandari, Won, Park and Park2018). Overall, there was less differentiation among the “sachalinensis-type” LEAFY sequences than among the “japonica-type” sequences: 43 nucleotide positions and 45 indels were variable among “japonica-type” sequences, whereas “sachalinensis-type” sequences had only 10 variable nucleotide positions and 5 indels.
In total, seven distinct multilocus genotypes were identified, when the matK and LEAFY data were considered together. Three accessions (R09, R12, R13) exhibited only “japonica-type” sequences, including three types of LEAFY sequence, and these were all recovered from Polk County in northwestern Wisconsin. Only “sachalinensis-type” sequences were recovered from two accessions from far northern Wisconsin (R24, R37), and two other accessions with missing matK data (R35, R37) may belong to this genotypic group as well. All other multilocus genotypic groups had the “japonica-type” matK or were missing matK data. Among these, the combination of “japonica-type 1” and “sachalinensis-type 1” LEAFY sequences was found in 11 individuals, mostly in southeastern Wisconsin (e.g., R01, R29). Ten individuals in northwestern and far northern Wisconsin (e.g., R17, R22) had the combination of “japonica-type 2” and “sachalinensis-type 2” LEAFY sequences. A small number of individuals (R06, R15, R26) had the “japonica-type 2” and “sachalinensis-type 1” LEAFY sequences, with no clear geographic pattern. Unique multilocus genotypes were recovered from R25 and R31, which each had the “sachalinensis-type 1” LEAFY sequence and two kinds of “japonica-type” LEAFY sequences.
Altogether, our data support the existence of genetically pure R. sachalinensis plants (i.e., without any evidence of R. japonica parentage) in northern Wisconsin, consistent with morphological identification of plants in that region. It is noteworthy, however, that not all of the plants with this genetic signature were identified as R. sachalinensis using morphological data, and the genetic composition and morphological variation of these plants merit further study. The R. japonica plants in our study with “japonica-type” matK sequences and only “japonica-type” LEAFY sequences are potentially genetically pure members of that species. Reynoutria japonica consists entirely of male-sterile individuals in the adventive range (Forman and Kesseli Reference Forman and Kesseli2003; Grimsby et al. Reference Grimsby, Tsirelson, Gammon and Kesseli2007; Hollingsworth and Bailey Reference Hollingsworth and Bailey2000), and the three accessions with exclusively “japonica-type” sequences (R09, R12, R13) were male-sterile. Although some genetically pure R. japonica or R. sachalinensis plants may exist in Wisconsin, the majority of plants we studied contained two kinds of LEAFY sequence, one kind attributable to R. japonica and one to R. sachalinensis. Five or more plants fell into each of the following categories. Plants with the J1 and S1 sequences were collected predominantly from southeastern Wisconsin (Figure 1). Plants with the J2 and S1 sequence combination were relatively rare but occurred evenly across the state. Northern and northwestern plants were largely characterized by the J2 and S2 combination. No plants were recovered with the J1 and S2 combination.
Plants with combinations of R. japonica and R. sachalinensis genetic elements are inferred to be hybrids. Hybrids with a male-sterile R. japonica ovule parent would be expected to have “japonica-type” matK sequences, and indeed we found this to be the case for suspected hybrid plants. The abundance of hybrid individuals in our study supports previous evidence that Reynoutria plants in North America have undergone extensive interbreeding (Gaskin et al. Reference Gaskin, Schwarzländer, Grevstad, Haverhals, Bourchier and Miller2014; Grimsby et al. Reference Grimsby, Tsirelson, Gammon and Kesseli2007). Similar genetic variability and phylogenetic reticulation have been reported for Reynoutria species in the native range (Park et al. Reference Park, Bhandari, Won, Park and Park2018). For example, three accessions from Honshu, Japan (j_hn03, j_hn04, and j_hn05), were found to have two different kinds of LEAFY sequence belonging to the “japonica-type” and “sachalinensis-type,” respectively (Figure 2), along with plastid parentage from either R. sachalinensis (j_h03) or R. japonica (j_h04 and j_h05; Figure 3). Therefore, the molecular tools have been established for verifying pure R. japonica or R. sachalinensis plants, as well as F1 hybrids and plants with intraspecific sequence variation, using a combination of nuclear and plastid sequence data.
Reynoutria taxa have a distinct ploidy in Polygonaceae, and their base chromosome number of x = 11 sets them apart from the related genera Fallopia and Muehlenbeckia that both have x = 10 (Schuster et al. Reference Schuster, Wilson and Kron2011). Moreover, in their native range, Reynoutria species have ploidy levels that range from tetraploid (2n = 44) to dodecaploid (2n = 132) (Park et al. Reference Park, Bhandari, Won, Park and Park2018). Ploidy is an important component of genetic diversity in plants, as polyploids often result from hybridization and sometimes become novel species (Doyle et al. Reference Doyle, Flagel, Paterson, Rapp, Soltis, Soltis and Wendel2008; Grant Reference Grant1981). Recently formed polyploids contain paralogous duplicates of nuclear genes, and over time the gene copies may be lost or become nonfunctional (Buggs et al. Reference Buggs, Doust, Tate, Koh, Soltis, Feltus, Paterson, Soltis and Soltis2009). Park et al. (Reference Park, Bhandari, Won, Park and Park2018) identified plants with apparently monomorphic LEAFY sequences, as well as plants having two or more copies of the LEAFY gene, with some copies resolving to distantly related clades on the LEAFY phylogeny (Figure 3). Some of the recovered copies of LEAFY may be allelic, and others could be paralogous in Reynoutria. We determined that most of the plants in Wisconsin have two distinct kinds of LEAFY sequence, most similar to reported sequences for R. japonica and R. sachalinensis, respectively. This pattern is consistent with interspecific hybridization, whereby the “japonica-type” and “sachalinensis-type” sequences would reflect the original parentage. Adventive Reynoutria populations have been characterized as tetraploid, hexaploid, or octaploid (Bailey Reference Bailey, Child, Brock, Prach, Pysek, Wade and Williamson2003; Bailey et al. Reference Bailey, Bímová and Mandák2007; Forman Reference Forman2003; Mandák et al. Reference Mandák, Pyšek, Lysák, Suda, Krahulcová and Bímová2003; Suda et al. Reference Suda, Trávníček, Mandák and Berchová-Bímová2010); thus, it might be possible to observe up to eight copies of the LEAFY gene in any one plant. We did not obtain chromosome numbers during this study, but the diversity of LEAFY genotypes we recovered is consistent with prior evidence that R. sachalinensis exists at lower ploidy levels than R. japonica and interspecific hybrids (Gammon et al. Reference Gammon, Baack, Orth and Kesseli2010). In future studies, it will be valuable to determine ploidy levels and compare these with genotypes to gain a more complete understanding of Reynoutria genetic diversity.
We collected morphological data for the plants that were studied, but we did not find a strong correlation between morphology and genotype, in contrast to what some studies have reported (e.g., Gaskin et al. Reference Gaskin, Schwarzländer, Grevstad, Haverhals, Bourchier and Miller2014). Only one plant (R37) was unambiguously scored as R. sachalinensis using both morphological and molecular methods. A large number of accessions were morphologically identifiable as R. japonica, but only three of these (R09, R12, R13) contained only “japonica-type” LEAFY sequences, and curiously these three accessions all had three kinds of LEAFY sequences, including the “japonica-type 4” sequence that was unique to them. The existence of plants that morphologically match R. japonica but contain “sachalinensis-type” genetic elements may indicate a need to reevaluate plants formerly identified as R. japonica. In the native range, plants identified as R. japonica also were found to have LEAFY sequences of both the “japonica-type” and the “sachalinensis-type” (j_hn03, j_hn04, j_hn05; Park et al. Reference Park, Bhandari, Won, Park and Park2018; Figure 3), confirming that genetic admixture between R. japonica and R. sachalinensis can complicate identification in both the native and invasive ranges. With updated knowledge about the widespread existence of hybridization and morphological variability, it will be important to evaluate the genetic and morphological diversity of populations in future studies.
It seems insufficient to characterize the invasive Reynoutria hybrids under a single taxon (i.e., R. × bohemica), because they contain such a wide array of genetic diversity and continue to produce later-generation hybrids. Many of the Reynoutria populations in Wisconsin, far from the original introduction sites for Reynoutria species in North America, likely represent later-generation hybrids, potentially with introgressed genetic material obtained through backcrosses with one or the other parental species. Our results are consistent with earlier evidence that R. × bohemica contains the greatest genetic diversity in the adventive range (Bailey Reference Bailey2013; Gaskin et al. Reference Gaskin, Schwarzländer, Grevstad, Haverhals, Bourchier and Miller2014; Hollingsworth et al. Reference Hollingsworth, Hollingsworth, Jenkins, Bailey and Ferris1998). High genetic diversity in invasive species could impact land management strategies, because diverse populations may be more capable of adapting to utilize novel habitats or evade control measures (Benoit et al. Reference Benoit, Les, King, Na, Chen and Tippery2019; Crawford and Whitney Reference Crawford and Whitney2010; Li et al. Reference Li, Ohadi and Mesgaran2020; Tippery et al. Reference Tippery, Pesch, Murphy and Bautzmann2020).
In this study, we determined that morphological characters may be insufficient for identifying Reynoutria plants in Wisconsin, and an ancillary study of molecular data would be advisable to help characterize individuals and populations. The DNA sequencing approach advocated here is relatively quick and inexpensive, and with the use of newly designed primers to distinguish “japonica-type” and “sachalinensis-type” LEAFY sequences, subcloning into bacterial vectors should not be required for most accessions. It will be valuable to expand the molecular evaluation of Reynoutria species across the adventive range to explore the possibility of multiple introductions from the native range, geographic patterns of diversity in North America, or other vital information that may help us to understand these pernicious plants.
Acknowledgments
No conflicts of interest have been declared. We are extraordinarily grateful to Maureen Ferry (Wisconsin Department of Natural Resources) for coordinating the collection of plant material and to the individual collectors (Shelby Adler, Katelin Anderson, Julie Benzschawel, Hannah Gargrave, Dan Hilger, Tyler Mesalk, Holly Schmaling, Alex Selle, Amanda Smith, Cassie Taplin, Andrew Teal, Matt Wallrath, Zach Wilson, and Eric Wojchik). Chong-Wook Park provided valuable input regarding DNA sequences. We thank Antonio DiTommaso for editorial assistance and two anonymous reviewers for their comments that helped to improve an earlier draft of the article. Funding was provided by the University of Wisconsin–Whitewater Undergraduate Research Program.




