


The first step in assessing relative effectiveness is an assessment of relative efficacy (1). To be granted regulatory approval, a new pharmaceutical has to be of sufficient quality, efficacy, and safety, with benefits outweighing the risks in a predefined patient population with a given disease. The efficacy and safety of a pharmaceutical is often demonstrated using disease-specific endpoint(s) in comparison to placebo and/or appropriate comparator(s) in premarketing clinical trials.
The subsequent assessment of the relative effectiveness and, if necessary, economic evaluation of a licensed pharmaceutical are necessary for determining its reimbursement status, access to market, and pricing. The relative effectiveness assessment (REA) of a pharmaceutical compares its benefits and harms in a target population to one or more alternative interventions (e.g., standard of care), evaluating if a new treatment has an added benefit (i.e., more benefit or less harm then a standard of care) (2) or is equivalent to existing alternatives. REA is based both on trials performed under ideal conditions (i.e., efficacy trials) and trials testing a pharmaceutical under conditions of everyday healthcare practice (i.e., effectiveness trials) (Reference Berkman, Lohr and Ansari3). REA is a component of health technology assessment (HTA). In addition to the REA, HTA includes ethical, legal, organizational, social, and cost-effectiveness considerations at the national or regional level. Reimbursement decisions are taken on the joint consideration of all the aspects of an HTA (Reference Berkman, Lohr and Ansari3). Quality of the evidence provided by the manufacturer on the REA needs to be adequate and sufficient; a pharmaceutical may not be accepted for reimbursement if the evidence provided by the manufacturer is considered inadequate by HTA agencies. Indeed, in addition to national healthcare priorities, available resources, utility of the technology to the healthcare system, the quality and adequacy of evidence is essential to support coverage decisions. In many cases, inadequate evidence regarding the benefits and harms of a pharmaceutical assessed is related to the choice of endpoints and/or comparator(s) used to demonstrate its benefits and harms.
Information regarding the procedure for reimbursement submission is available in most European HTA agencies; however, guidelines for the methodology to apply for the REA are scarce (Reference Kleijnen, George and Goulden4). Therefore, one of the important aims of the European collaboration in the field of HTA, led by a voluntary European Network of Health Technology Assessment (EUnetHTA), has been to elaborate methodological guidelines on main issues related to the REA of pharmaceuticals. The purpose of the guideline development process was to establish a framework for appropriate methodology of REA that can be applied by health technology assessors across countries in Europe. This could improve consistency in assessment across HTA bodies, industry and decision makers.
The aim of this article is to describe the guideline development process, and the key findings with regards to the use of endpoints for REA of pharmaceuticals and the recommendations that resulted from it.
METHODS
Guidelines Development Process
The development of guidelines was one of the deliverables of the European Network of Health Technology Assessment (EUnetHTA) JA1 Work Package (WP) 5, led by College voor Zorgverzekeringen (CVZ), Netherlands, and co-lead by Haute Autorité de Santé (HAS), France. Seventeen associated and twelve collaborative EUnetHTA partners participated in guidelines development. HAS was specifically responsible for the coordination of guidelines elaboration within WP5, notably coordination of timelines and meetings, review of successive drafts and received comments, as well as assuring consistency between inter-related guidelines and corresponding recommendations.
Identification of Topics
The first step in the elaboration of guidelines decided by the EUnetHTA partners was to identify the most important concepts, to agree on the terminology to use throughout the process, for example, relative efficacy and effectiveness, definition of a comparator and of endpoints of interest for REA, and to choose the most relevant methodology issues for REA. The three key domains relevant for REA were considered to be: endpoints, comparators/comparisons, and levels of evidence. The key topics identified for these domains gave rise to nine guidelines: endpoints (clinical, surrogate, composite, and health-related quality of life endpoints), safety, comparators and comparisons (appropriate comparators, direct and indirect comparisons); and levels of evidence (internal validity of randomized controlled trials, applicability of evidence).
Authors’ Selection and Literature Search
Once the topics and general guideline structure were agreed upon by all WP5 partners, authors were selected on a voluntary basis, with one or two authors per guideline. The authors first prepared a scoping document to be approved by the EUnetHTA JA1 WP5 Lead and Co-Lead before completing a literature search with predefined key words relevant to the guideline topic; the literature search was supplemented by the authors of this article (last search: end 2013) with the HTA assessors’ experience on endpoints use from several applications submitted for REA.
Internal and External Consultations
The internal and external consultations of draft guidelines began in January 2011 (Figure 1). Final versions of guidelines were completed in March 2013.
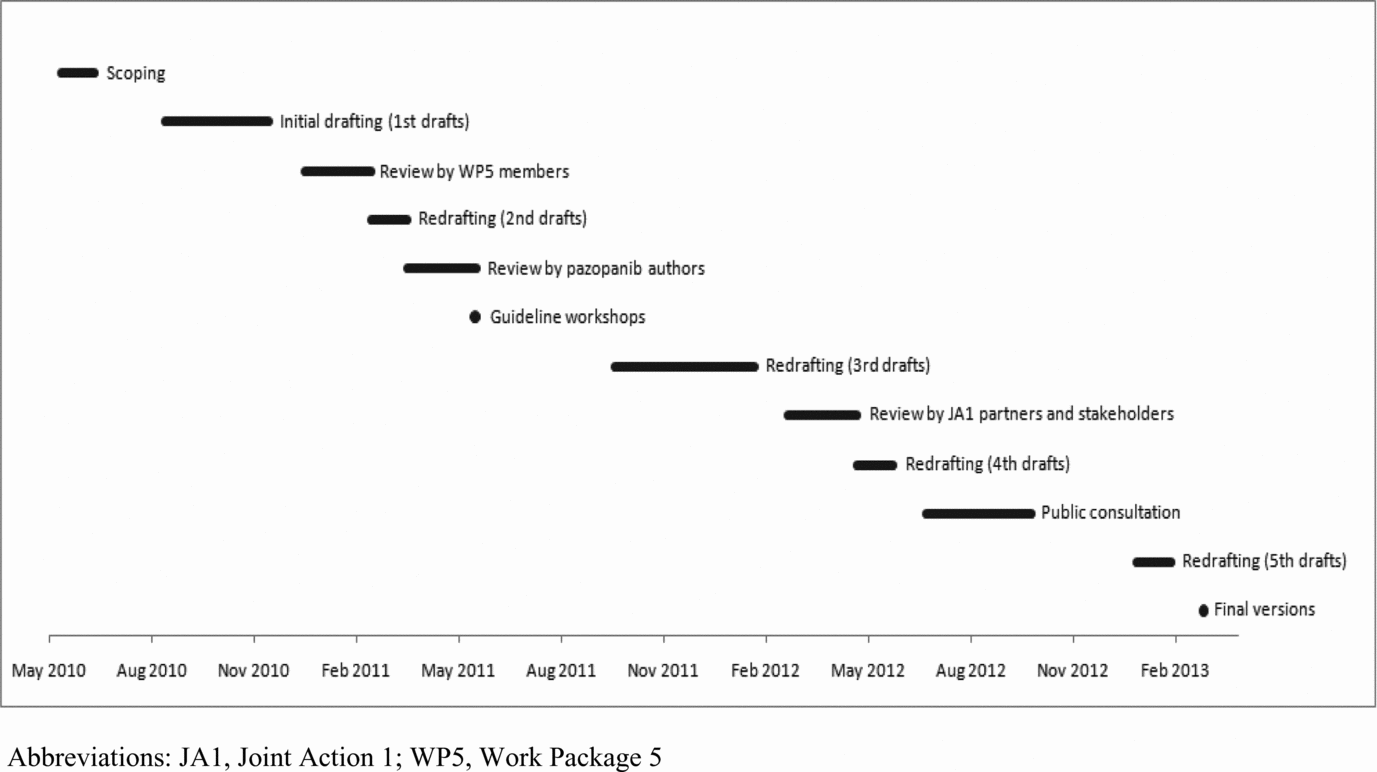
Figure 1. Guidelines development timelines and review process.
Electronic and Face-to-Face Meetings
In addition to electronic exchanges, several face-to-face meetings were organized to discuss the most relevant and/or contradictory issues, both internally (with some or all WP5 members) and externally, with industry and EUnetHTA Stakeholder Forum representatives.
RESULTS
Guidelines Development Process
After the first internal consultation of the EUnetHTA partners, authors received between 115 and 170 comments per guideline. The majority of comments were editorial in nature. However, comments were also received on the structure, scope, terminology, definitions of concepts used, and the necessity to give clear recommendations for assessors conducting REA. The second and third consultations were of a similar nature. The main issues were identified and thoroughly discussed by guideline authors, the coordinating agency (HAS) and EUnetHTA partners. The most relevant comments from the EUnetHTA partners were introduced in the fourth drafts of guidelines that were sent out to public consultation.
The number of comments received after public consultation was similar to that received during internal consultation (100 to 160 per guideline); most were related to the proposed methodology framework for the REA, and the content and relevance of the recommendations given. The final guidelines content was the result of the consideration of all comments received and the consensus reached by EUnetHTA partners on the key issues relating to REA methodology. Final guidelines and tables with all received comments and authors’ answers were made available for consultation at the EUnetHTA website.
Endpoints Categories for REA
Clinical Endpoints
Clinical endpoints is the umbrella term for patient-relevant endpoints used in REA that describe how a patient feels, functions or survives (5).
Clinical endpoint should be a valid measure of clinical benefit or harm due to treatment (6). Therefore, it should be well-defined and justified, relevant, responsive to changes due to treatment, reflect evolution of a disease, reproducible, free of measurement or assessment error, and unbiased (6).
Clinical endpoints can be broadly divided in three main categories: mortality, morbidity (due to disease or its treatment) and health-related quality of life (HRQoL). Definitions and examples of endpoint categories are given in Tables 1 and 2.
Table 1. Endpoints Definitions

Table 2. Examples of Endpoints

Clinical or patient-relevant endpoints may be reported by a patient (Patient-Reported Outcomes), a clinician, a caregiver or an observer (e.g., pediatrics) (6). Patient-relevant endpoints should not be confused with patient-reported outcomes, even if some patient-relevant endpoints are reported by patients themselves.
Patient-reported outcomes (PRO) is an umbrella term used to describe any outcome evaluated directly by the patient himself/herself and based on patients’ perception of a disease and its treatment(s) (7–9). HRQoL represents a specific type/subset of PROs, distinguished by its multi-dimensionality (7;9).
In general, trial endpoints can be symptoms such as pain, dyspnea, or anxiety, final mortality or morbidity endpoints such as myocardial infarction, stroke, or fracture, surrogate endpoints such as HIV viral load, blood pressure, HbA1c, or intermediate endpoints such as progression-free survival, or angina frequency (6). All may be used either as single endpoints or as composite endpoints (i.e., two or more endpoints combined into one endpoint) if adequate to measure clinical benefit (6).
Surrogate Endpoints
The acceptability of an endpoint as a surrogate for a specific clinical endpoint is based on its biological plausibility and empirical evidence (validation) (5;10–13). An REA should, whenever possible, be based on final patient-relevant clinical endpoints (13). However, surrogate endpoints could be accepted in the initial assessment if the validity of the surrogate/final clinical endpoint relationship has been clearly established a priori (Reference Fleming and De Mets11;13), even if trials based on surrogate primary endpoints are more likely to report larger treatment effects than trials reporting final patient relevant primary endpoints (Reference Ciani, Buyse and Garside14). For the re-assessment of a pharmaceutical, effectiveness is recommended to be demonstrated on final clinical morbidity and mortality endpoints (13). It is important at this stage that there are sufficient safety data.
The absence of data on clinical endpoints relevant for REA might be acceptable when a clinical endpoint is difficult or impossible to study (e.g., very rare or delayed) or the target population is too small to obtain meaningful results on relevant clinical endpoints even after very long follow-up (e.g., very slowly progressing and/or rare diseases) (13).
Composite Endpoints
The use of composite endpoints should be avoided if suitable single endpoints are available (15). The components of a composite endpoint should be of similar clinical importance and be limited to three or four. The prior empirical evidence of the value of each component endpoint must be defined. The contribution of each component to the result within the composite endpoint should be reported. All components should also be reported separately as secondary endpoint according to the endpoint hierarchy (15). Assessors need to be able to discern the effect of the intervention on all components of a composite endpoint. If a significant difference is obtained on the composite endpoint, but the effect is not homogeneous across components, it cannot be concluded that the treatment has an effect on the composite endpoint as a whole (15).
HRQoL Endpoints
The impact of an intervention on HRQoL should be systematically assessed in REA if appropriate and sufficient data are available. Along with mortality and morbidity, HRQoL is one of the major REA endpoints (9). The improvement in HRQoL alone (for equivalent effectiveness and/or harms) may be the basis for “added benefit” of a new drug compared with an adequate comparator.
HRQoL is typically measured using a validated instrument. The appropriateness of the HRQoL measure used depends on the purpose of the REA. If REA is used to inform healthcare policy makers about the relative value of a product, the decision-making context plays a crucial role (9). In a context where drug reimbursement decisions take into account cost-effectiveness data as well as priority setting values across indications, generic HRQoL measures that translate into utility values are preferred to disease-specific HRQoL measures to maximize the comparability of REAs across indications (9). Disease-specific measures may be added as complementary information. When cost-effectiveness is considered within indications only, disease-specific HRQoL is in principle sufficient, although adding a generic utility measure is useful for coherence across decisions (9).
DISCUSSION
The choice of endpoints to demonstrate benefits and acceptable harms of a pharmaceutical, and the manner in which they are reported, can have a major impact on regulatory decisions (16). With regards to the REA for reimbursement purposes, endpoints deemed inadequate or inadequately reported (Reference Wieseler, McGauran and Kaiser17) by health technology assessors are an important reason for poor decision making.
The EUnetHTA guidelines recommend that the choice of endpoints depend on the target population studied, the characteristics of the disease and its core symptoms and signs, as well as the intended purpose of treatment, that is, diagnostic, preventive, curative, symptomatic, palliative, the unintended side effects (6) and the decision making context, for example, whether or not cost-effectiveness information is used for deciding on reimbursement. In the context of the REA of pharmaceuticals, preference is clearly given to long-term or final endpoints whenever possible (6), as opposed to short-term endpoints that may only be acceptable for acute symptomatic conditions with no long-term consequences. Surrogate endpoints are accepted when they are validated, that is, when there is compelling evidence of a clear and consistent correlation between the effect of treatment on the surrogate and the effect on the final outcome of interest (5;10–Reference Ciani, Buyse and Garside14). Surrogate endpoints are of specific importance when long-term or final endpoints are difficult to measure (e.g., slow progression, rare disease).
The relevance and hierarchy of the different clinical endpoints will depend on the research question, the disease, and on the treatment investigated. However, even if a trial is powered on a primary endpoint, the added clinical benefit of a new pharmaceutical will be assessed in comparison to an adequate comparator on all endpoints relevant for a disease or its treatment (6). In addition, a generic endpoint such as life years gained or quality-adjusted life years gained is often considered to enhance the relevance of the REA for the decision making process in its broad context (9). Adjustment for multiple hypothesis testing may be appropriate. This simultaneous assessment of all relevant endpoints appears as a hallmark of REA.
The EUnetHTA guidelines on endpoints for REA, summarized in this article, are aimed at providing clarity in this field and support both health technology assessors in preparing the REA and the industry in planning an adequate development as they describe the type of data required for REA. Since the finalization of guidelines, the EUnetHTA partners report their increased use both in countries lacking national methodological guidelines and in countries with available ones (18).
The guidelines development process was complicated, time and resources consuming, both for guideline authors and the coordinating agency. As reimbursement rules differ across the countries that participated in this task, it was not easy to identify and methodologically articulate the common approach to key issues that can be applied beyond and above the national rules and procedures. Indeed, consensus on key issues was probably the most difficult and the most important goal achieved. However, the iterative process used with several drafts of each guideline, each of which was reviewed, and the large number of internal and external reviewers helped ensure quality of content and high degree of consensus reached and reflected in finalized guidelines.
The existing guidelines on the methodology of REA of pharmaceuticals might need to be revised once first experiences with their use become available from on-going EUnetHTA pilot projects. Currently, based on the experience gained, the process of the development of methodology guidelines is being refined and enlarged to cover other important issues for HTA such as internal validity of nonrandomized clinical trials, meta-analysis of diagnostic test accuracy studies and economic evaluation.
In addition to general methodology guidelines, in the framework of the EUnetHTA Joint Action 2, there is on-going work on a disease-specific guideline covering specific aspects of a technology development for particular disease and conditions. In addition, multi-HTA early dialogues with health technology developers on the evidence to provide during health technology development including adequate choice of patients, endpoints, comparators and comparisons, are part of the current EUnetHTA work. These activities aim to improve the initial evidence generation for HTA, optimize health technology development and support adequate and timely coverage decisions for a given product and condition.
CONCLUSIONS
The choice of endpoints relevant for REA depends on the disease, population, treatment and decision context. Not only the primary endpoint of a study, but also other relevant endpoints should systematically be assessed and should always reflect how a patient functions, feels, and survives. The simultaneous assessment of all relevant endpoints is a hallmark of REA, as the added clinical benefit of a new pharmaceutical is assessed in comparison to an adequate comparator on all endpoints considered relevant for a disease or its treatment. While assessing HRQoL, generic utility measures should be considered and complemented with disease-specific measures. Long-term or final endpoints are preferred whenever adequate or feasible; short-term endpoints are adequate for acute conditions with no long-term consequences and as measures of relevant symptoms of a disease. Surrogate endpoints may be used when they are validated against the final outcome of interest.
CONTACT INFORMATION
Mira Pavlovic, MD (m.pavlovic@has-sante.fr), La Haute Autorité de Santé 2, avenue du Stade de France 93218 Saint-Denis La Plaine Cedex, Saint-Denis la Plaine 93218, France
Conor Teljeur, BE, MSc, PhD, Health Information and Quality Authority, Dublin, Ireland
Beate Wieseler, MSc, PhD, Institute for Quality and Efficiency in Health Care, Cologne, Germany
Marianne Klemp, MD, PhD, Professor, Norwegian Knowledge Centre for the Health Services, Oslo, Norway
Irina Cleemput, PhD, Professor, Belgian Health Care Knowledge Centre, Brussels, Belgium; Hasselt University
Mattias Neyt, MSc, PhD, Belgian Health Care Knowledge Centre, Brussels, Belgium
CONFLICTS OF INTEREST
All authors report their institutions received funding from the European Commission Grant to support the EUnetHTA Joint Action 1 tasks related to the relative effectiveness assessment of pharmaceuticals. No other conflicts of interest have been declared.



