


Introduction
The search for evidence of life on Mars will rely on the detection of biomolecules and universal markers (Fajardo-Cavazos et al. Reference Fajardo-Cavazos, Schuerger and Nicholson2010). Due to their unique role in storing the genetic information of all forms of life on Earth, nucleic acids, specifically DNA, have been considered a ‘biosignature’ of life (Lyon et al. Reference Lyon, Monier, Dupraz, Freissinet, Simonet and Vogel2010). DNA is able to attach and bind to mineral surfaces (Cleaves et al. Reference Cleaves, Jonsson, Jonsson, Sverjensky and Hazen2010) and survive strong UV exposure whilst retaining biological activity if bound to surfaces in plasmid form (Lyon et al. Reference Lyon, Monier, Dupraz, Freissinet, Simonet and Vogel2010). Additionally, recent studies indicate that forward-contaminant DNA could exist for considerable periods of time on the Martian surface (Fajardo-Cavazos et al. Reference Fajardo-Cavazos, Schuerger and Nicholson2010).
Due to significant meteoritic exchanges between Mars and Earth, there is a reasonable chance that potential past or present life on Mars could be related to life on Earth. Impact-induced ejections of rocks from planetary surfaces were frequent events in the early history of the solar system and have been considered possible first steps in the potential interplanetary transfer of micro-organisms (Moeller et al. Reference Moeller, Horneck, Rabbow, Reitz, Meyer, Hornemann and Stöffler2008). Thus, the search for DNA on Mars could lead to a significant insight: if potential Martian life is based on DNA, this would strongly suggest a common origin of life on Earth and Mars. For this search, supremely sensitive technologies used to study life on Earth, including life in extreme environments, can be applied to the search for life on other planets (Isenbarger et al. Reference Isenbarger, Carr, Johnson, Finney, Church, Gilbert, Zuber and Ruvkun2008). Interestingly, identification of bacterial communities by DNA extraction has already been successfully performed in other extreme conditions such as Australian hypersaline environments with extremely low pH (Mormile et al. Reference Mormile, Hong and Benison2009), the Antarctic Dry Valleys which include some of the coldest, driest and most oligotrophic soils (Cary et al. Reference Cary, McDonald, Barrett and Cowan2010) and the Atacama Desert of northern Chile which is one of the most arid deserts on Earth (Drees et al. Reference Drees, Neilson, Betancourt, Quade, Henderson, Pryor and Maier2006; Connon et al. Reference Connon, Lester, Shafaat, Obenhuber and Ponce2007).
In order to assess several human and scientific aspects of future robotic and manned missions on planetary surfaces, the EuroGeoMars campaign was performed as part of the ExoGeoLab pilot project developed at ESTEC/ESA (European Space Agency) in collaboration with ILEWG, NASA Ames and European and US investigators (Foing Reference Foing2009; Ehrenfreund et al. Reference Ehrenfreund, Foing, Stoker, Zavaleta, Quinn, Blake, Martins, Sephton, Becker and Orzechowska2010; Foing et al. Reference Foing, Mahapatra, Boche-Sauvan, Som, Page, Stoker, Zhavaleta, Sarrazin, Blake and Poulakis2010; Ehrenfreund et al. Reference Ehrenfreund, Röling, Thiel, Quinn, Septhon, Stoker, Direito, Kotler, Martins, Orzechowska, Kidd and Foing2011; Foing et al. Reference Foing, Stoker, Zavaleta, Ehrenfreund, Thiel, Sarrazin, Blake, Page, Pletser, Hendrikse, Direito, Kotler, Martins, Orzechowska, Gross, Wendt, Clarke, Borst, Peters, Wilhelm and Davies2011) at the Mars Desert Research Station. The MDRS is a Mars analogue research station (Fig. 1(a)), situated in the San Rafael Swell in Utah, about 7 miles from Hanksville. The primary goal of our study was to test the possibility of analysing soil samples for microbial life, on-site in the habitat's laboratory. We wanted to establish a routine sample analysis for the detection of microbial DNA based on the polymerase chain reaction (PCR) technique, going far beyond the techniques that other MDRS crews had previously used, such as microscopic analysis or the conventional cultivation of microbial samples on agar or other nutrient media (MDRS crews 1b, 7, 11, 44, 52 in Secosky Reference Secosky2008). The PCR is an effective tool to identify trace amounts of DNA, ideally down to the level of a single molecule (Saiki et al. Reference Saiki, Scharf, Faloona, Mullis, Horn, Erlich and Arnheim1985; Mullis & Faloona Reference Mullis and Faloona1987; Sermon & De Rycke Reference Sermon and De Rycke2007). The establishment of such a technique would be a powerful tool for the detection of traces of life, since non-culturable micro-organisms or even endospores could be detected as well.

Fig. 1. The Mars Desert Research Station. (a) The habitat, the green house and the All Terrain Vehicles are shown (Credit: The Mars Society). (b) The newly installed molecular biology laboratory at MDRS. Vortex, balance, centrifuge, PCR machine and glove box are shown (Credit: EuroGeoMars crew 77). (c) First floor of the MDRS habitat. The molecular biology working area is highlighted in red (Credit: The Mars Society).
Soils and sediments often contain DNA that can be amplified by PCR (Hofreiter et al. Reference Hofreiter, Mead, Martin and Poinar2003; Willerslev et al. Reference Willerslev, Hansen, Binladen, Brand, Gilbert, Shapiro, Bunce, Wiuf, Gilichinsky and Cooper2003) and bacterial DNA sequences have been found in sediments that go back over half a million years (Willerslev et al. Reference Willerslev, Hansen, Ronn, Brand, Barnes, Wiuf, Gilichinsky and Cooper2004). Therefore, there is the possibility that DNA bound to minerals (Cleaves et al. Reference Cleaves, Jonsson, Jonsson, Sverjensky and Hazen2010; Lyon et al. Reference Lyon, Monier, Dupraz, Freissinet, Simonet and Vogel2010) could exist on the Martian surface (Fajardo-Cavazos et al. Reference Fajardo-Cavazos, Schuerger and Nicholson2010) and be accessible for analysis. For the ultimate goal of analysing soil samples from Mars on-site, a sensitive, robust and reliable detection method has to be developed, optimized and tested under Mars analogue conditions.
Material and methods
Instruments for the PCR experiment at MDRS
All instruments and consumables were evaluated for their usability in biological experiments at MDRS in pre-mission tests in a conventional laboratory as well as at ESTEC in The Netherlands. The following instruments were shipped to MDRS: a precision balance (Satorius, Germany) used to weigh 0.25 g of soil sample for the DNA purification, a Vortex (vortex genie2, Fisher Scientific, Germany) and a centrifuge (EPP cent mini spin plus, Eppendorf, Germany) required for DNA extraction. A glove box (self-built) was used to pipette the PCR mixtures. Fragment amplification from sample DNA was performed in the thermal cycler Primus 25 advanced (Peqlab, Germany). PCR fragments were separated according to their molecular weights and viewed with 1.2% agarose E-Gels containing SYBRsafe (Invitrogen, Germany) using the E-Gel iBase system (Invitrogen, Germany) and viewed with the E-Gel Safe Imager Real-time Transilluminator (Invitrogen, Germany). Agarose gels were documented with a digital camera (Coolpix 995, Nikon) on a tripod.
Sample collection
It was decided before the mission that samples would be collected at different depths (0 to −2, −10, −30 cm) in places both without and near vegetation in order to compare the microbial content. Samples were intended to be taken by drilling and soil should have been taken from the drilling core by measuring the desired depths. Unfortunately, it became clear during the mission that the soil was too dry and it was not possible to obtain an intact drilling core. Additionally, sampling down to −30 cm was often not possible because of the stony or solid-clay soil structure. Therefore, soil samples were collected with a shovel, and sterile soil sampling spatulas. After arriving at the chosen sampling location (Table 1), the ground was examined and a location without any visible human or animal traces was chosen. We followed this strategy because we did not want to have false positive micro-organisms in our samples that would invalidate the analysis of the microbial communities’ content. For the sampling, all collecting instruments (shovel, soil-sampling spatula) were sterilized by wiping them with 70% ethanol solution to remove any contamination. Surface samples (0 to −2 cm depth) were directly collected with a sterile spatula. For sampling depths of −10 and −30 cm, a hole was dug with a sterilized shovel and the soil samples were taken from the side of the hole with a sterile spatula to avoid contamination from the upper soil layers. Samples were transferred into sterile sampling bags (VWR International, Germany) and stored until return to the habitat laboratory. The shovel was wiped with 70% ethanol between the different sampling steps and for each sampling a new, sterile spatula was used.
Table 1. Description of the sites chosen for collection of soil samples

<d.l.: DNA concentration below detection limit.
DNA extraction methods
The DNA extraction was performed using the PowerSoil DNA Isolation Kit (MOBIO Laboratories, USA). This extraction method allows genomic DNA to be isolated from environmental samples containing a high humic acid content, which usually impairs DNA quality and hinders subsequent enzymatic reactions such as PCR amplification of microbial DNA. According to the manufacturer, this kit can be used to extract DNA from various soil types, such as compost, sediment and manure.
The DNA was extracted from the soil samples as follows: 0.25 g of soil, accurately measured with a scale, was taken from the collected soil in the sterile sampling bag and added to the supplied bead-beating tube before being mixed by vortexing. Then 60 μl of the lysis solution C1 was added to the tube and the mixture was briefly vortexed. The sample tube was taped to the flat-bed vortex and vortexed at maximum speed for 10 minutes. Thereafter, the tube was centrifuged at 10 000 g for 30 seconds at room temperature. The supernatant was transferred to a clean 2 ml collection tube. Then 250 μl of solution C2 was added to precipitate the non-DNA organic and inorganic material including humic substances, cell debris and proteins. After vortexing the solution for 5 seconds and incubation for 5 minutes at 4°C, the tube was centrifuged at room temperature for 1 minute at 10 000 g. A maximum volume of 600 μl of supernatant was transferred into a clean collection tube. Then 200 μl of C3 solution, a second reagent to precipitate additional non-DNA organic and inorganic material, was added and the mixture was incubated at 4°C for 5 minutes. After centrifugation for 1 minute at room temperature at 10 000 g, a maximum volume of 750 μl of supernatant was transferred into a clean collection tube. The solution was mixed with 1200 μl of C4, a high salt solution, and vortexed for 5 seconds. 675 μl of this solution was then loaded onto a spin filter and centrifuged for 1 minute at 10 000 g at room temperature to bind the DNA to the silica material. The rest of the solution was loaded onto the membrane in a second centrifugation step. 500 μl of the ethanol-based wash solution C5 was pipetted onto the silica membrane, the tube was centrifuged for 30 seconds at 10 000 g and the flow through was discarded. After a second centrifugation step, the silica membrane was transferred to a clean collection tube and 100 μl of elution buffer C6 (10 mM TrisCl, pH 8) was added to the centre of the membrane. The DNA was eluted by centrifugation at room temperature for 30 seconds at 10 000 g and then stored in the freezer compartment of the laboratory fridge until it was used for PCR. (For a detailed DNA extraction protocol, including buffer C1–C6 application, see the manufacturer's manual: http://www.mobio.com/images/custom/file/protocol/12888.pdf).
In order to test for potential sample contamination by microbial or human DNA due to dust and airflow in the MDRS laboratory, the DNA extraction procedure was also performed with 0.25 g of sterile water instead of soil and the eluate was used as a negative control for PCR analysis.
Quantification of DNA content in collected soil samples
The amount of DNA after purification with the PowerSoil DNA Isolation Kit (MOBIO Laboratories, USA) was analysed by using the Qubit dsDNA HS assay kit in combination with the Qubit Fluorometer (Invitrogen, Germany). The assay uses the fluorescent dye PicoGreen that binds to double stranded (ds)DNA leading to an enhancement in fluorescence. This method is up to 1000 times as sensitive as UV absorbance readings and therefore suitable for very low sample concentrations (1–500 ng ml−1). Moreover, the measurement is unaffected by many contaminants such as free nucleotides, salts, solvents and proteins. The measurement was performed according to the manufacturer's protocol (http://probes.invitrogen.com/media/pis/mp32851.pdf). In brief, 10 μl of the supplied standard solutions or the purified DNA samples was mixed with 190 μl of the Qubit working solution (dsDNA HS reagent:Qubit dsDNA HS buffer, 1:200). The reactions were incubated at room temperature for 2 minutes before measurement of fluorescence with the Qubit Fluorometer and calculation of DNA concentration (Table 1).
PCR fragment amplification and visualization
DNA extracted from the soil samples was used in PCR experiments with specific primers for each of the three domains: bacteria, archaea and eukarya (fungi). One μl of the DNA extracted from the 0.25 g of soil with the PowerSoil DNA isolation kit was used per PCR mixture (consisting of: specific primers (Table 2) and Taq PCR MasterMix Kit components (Qiagen, Germany)). PCR amplifications were performed with this basic protocol: initial denaturation at 94 °C for 5 minutes, 30 cycles of denaturation at 94°C for 1 minute, annealing at variable temperatures (see Table 2) for 45 seconds, elongation at 72°C for variable times (see Table 2), final-elongation at 72 °C for 10 minutes. In order to prevent cross-contamination with micro-organisms from room air or raised dust, a glove box was used to prepare the PCRs. The DNA was amplified in 30 cycles using a Primus25 advanced cycler (PeqLab, Germany) with optimized parameters for the different primer sets (Table 2). Afterwards, the PCR samples (25 μl) were applied onto a 1.2% agarose gel containing the SYBRsafe DNA gel stain dye (E-Gel 1.2% with SYBRsafe, Invitrogen, Germany) next to a molecular weight marker as a fragment length standard (TRACKIT 100 bp DNA ladder, Invitrogen, Germany) and run for 26 minutes in the E-Gel iBase system (Invitrogen, Germany). A digital picture (Coolpix 995, Nikon) of the gel was taken on the E-Gel Safe Imager Real-time Transilluminator (Invitrogen, Germany) under the amber filter (blue light filter for eye protection).
Table 2. PCR primers and conditions

After the mission, all of the collected samples were re-analyzed using the PCR technique in a laboratory at Mesa State College in Grand Junction, Colorado, using the same PCR mixture components and PCR programmes as for the MDRS experiments. Unfortunately, the agarose gel visualization system had to remain in the laboratory at MDRS and the agarose gel preparation, staining and documentation of the sample re-analysis in the college laboratory had to be performed differently. The PCR samples were run on a 1% unstained agarose gel in TAE buffer. A 1 kb DNA ladder (1 kb Plus DNA Ladder Fermentas, Germany) was used as a molecular weight marker. The gel was run for 30 minutes and an ethidium bromide containing card (InstaStain Ethidium Bromide, EDVOTEK, USA; http://www.edvotek.com/pdf/instastain_ethidium.pdf), was placed onto the gel for 2–4 minutes to stain the DNA. Although this is not the standard method for staining weak amplification products, it was the routine staining method in the Mesa College Laboratory. The gels were placed on an UV-transilluminator and documented with an imaging system.
Results
Pre-mission tests for instrument selection
The biology experiment of MDRS crew 77 was tested beforehand in the home laboratory of one of the authors to select the most adequate instruments and techniques for identifying microbial DNA by PCR. The optimal PCR conditions were analysed for the different primer sets specific for bacteria, fungi and archaea. Optimal PCR fragment amplification conditions such as annealing temperature, elongation time and cycle number were identified (Table 2) using Escherichia coli DNA or microbial DNA isolated from garden soil (Fig. 2) as positive controls for the PCR (Fig. 3). A PCR with bacterial primers was performed on the DNA extracted from 2 MDRS test samples to complete the pre-mission test at ESTEC (Fig. 3). All necessary instruments and consumables were tested for their versatility and easiness of future use in a small laboratory such as the one at MDRS. Emphasis was placed on easy handling, a limited number of procedure steps, non-toxicity and a minimum amount of working space. The experimental equipment was transported to ESTEC in the Netherlands for the final pre-mission test (Fig. 3) and shipped to MDRS to be installed and functionally tested in the laboratory by the technical crew and crews 76 and 77 (Fig. 1(b) and (c)).

Fig. 2. Detection of DNA extracted from a soil sample by the PowerSoil DNA Isolation Kit. 1, 4: commercially molecular weight marker as a fragment length standard (1 kb Plus DNA Ladder; the 20 kilo base pairs (kb) fragment is indicated); 2, 3: 10 μl of purified DNA from a compost soil sample.

Fig. 3. Pre-mission PCR test. Reaction conditions were optimized for the identification of bacterial DNA in the collected soil samples. (a) Agarose gel showing the amplified DNA fragments of the pre-mission test performed with the garden soil sample. 1: Commercially available molecular weight marker as a fragment length standard (1 kb Plus DNA Ladder; the 1.5 kb fragment is indicated); 2: negative control containing all reaction components but no DNA; 3: DNA extracted from garden soil sample; 4: positive control containing E. coli DNA 2–4: 25 μl of the PCR mixture was applied to the agarose gel. (b) Agarose gel showing the amplified DNA fragments of the pre-mission test performed with the soil samples collected at MDRS. 1: Commercially available molecular weight marker as a fragment length standard (1 kb Plus DNA Ladder; the 1.5 kb fragment is indicated); 2: negative control containing all reaction components but no DNA; 3: DNA extracted from MDRS test soil sample 1; 4: DNA extracted from MDRS test soil sample 2; 5: positive control containing E. coli DNA 2–5: 25 μl of the PCR mixture was applied to the agarose gel.
Soil sampling
In total, 25 soil samples were collected (Table 1). The soil sample collection sites can be divided into two categories: (i) without vegetation (Fig. 4) and (ii) close to sparse vegetation that consisted of small bushes or grass (Fig. 5). Additionally, 5 water samples were collected and microbial DNA was extracted to test the versatility of the experimental set-up for future missions within a liquid sample setting (Table 1, Fig. 6).

Fig. 4. Examples of soil sampling locations without neighbouring vegetation (Credit: EuroGeoMars crew 77).

Fig. 5. Examples of soil sampling locations with neighbouring vegetation (Credit: EuroGeoMars crew 77).

Fig. 6. Examples for water sampling locations (Credit: EuroGeoMars crew 77).
DNA extraction and PCR
Microbial DNA was immediately isolated from the soil samples with the PowerSoil DNA Isolation Kit after return to the MDRS laboratory and stored in the freezer compartment of the laboratory fridge until PCR analysis. Long-term storage of collected soil samples was avoided because this would modify growth conditions and could impair the viability of some micro-organisms, leading to a change in microbial content. Because of the different requirements of the scientists who were working at MDRS laboratory working hours were divided. While geologists needed to crush their samples, e.g. for X-ray diffraction spectroscopy, which produced a lot of dust, molecular microbiology analysis needed a clean and dust-free working area. In particular, the PCR is a highly sensitive technique and the smallest traces of contamination lead to false positive results. This technical challenge was addressed by defining alternating working hours for the scientist groups, including negative controls in the experiments, as well as the usage of a glove box (Fig. 1(b)) to prevent contamination during pipetting of the PCR components. PCR runs were performed according to the optimized reaction conditions (Table 2). The amplified DNA fragments were visualized directly after the PCR by using agarose gels with SYBRsafe staining and a digital imaging for recording. All the PCR experiments performed at MDRS are shown in Fig. 7 and summarized in Table 3. There was no cross-contamination detectable, which, would have been immediately visible in the negative control included for each PCR experiment as well as in the negative control prepared to monitor the DNA purification. As experiment time was limited due to various other habitat maintenance chores that had to be performed (Thiel et al. Reference Thiel, Pletser and Foing2011), only 26 out of 30 samples were analysed by PCR for their bacterial and fungal content at MDRS (Fig. 7). The remaining 4 samples were analysed the day after the mission in a laboratory at Mesa State College in Grand Junction, Colorado. In total, it was possible to detect bacterial DNA in 14 and fungal DNA in 8 of the 26 analysed samples.

Fig. 7. PCR experiments performed at MDRS. Upper part: PCR with bacterial primers. Lower part: PCR with primers specific for fungi. 01–26: PCR amplification of DNA from collected samples. pc: positive control: DNA extracted from E. coli. nc: negative control: reaction with water substituting the DNA. m: commercially available molecular weight marker as a fragment length standard (TrackIT 100 bp DNA ladder; filled arrows indicate the 1.5 kb fragment; open arrows indicate the 0.6 kb fragment). For each PCR sample 25 μl of the reaction mixture was applied to the agarose gel.
Table 3. Results for bacteria, eukarya and archaea rRNA PCR analyses performed at MDRS and Mesa College Laboratory

+: positive signal; −: no signal; n.a.: not analysed.
The concentration of the DNA extracted from the soil samples was analysed in the home laboratory of one of the authors after the mission (Table 1). Due to the expected low levels of DNA content, a technique using a DNA-binding fluorescent dye was applied, which enhances the sensitivity by about 1000-fold compared to UV absorbance measurements (see Material and methods section). DNA concentration ranged from 0.018 to 17.6 μg ml−1. It is remarkable that despite the very low DNA concentration, which was below the detection limit in about 50% of the samples (see Table 1), it was still possible to detect the presence of micro-organisms with this established PCR assay.
PCR analysis quality at MDRS
Directly after the mission, all samples were re-analysed in a conventional laboratory setting at Mesa State College using the same bacterial, fungal and archaeal primers such as at MDRS (Fig. 8, Table 3). The direct comparison of PCR results for samples 1–16 performed at MDRS and at the Mesa College Laboratory showed that 14 of the 16 MDRS PCR samples (87.5%) contained bacterial DNA, while 8 samples (50%) contained fungal DNA. Slightly different results were obtained for PCR results performed at the Mesa College Laboratory: in 10 samples (62.5%) bacterial DNA and in 8 samples (50%) fungal DNA could be detected. In 4 of the samples, positive in the analysis at MDRS using the bacterial primers, no PCR product was detected in the re-analysis at the Mesa College Laboratory (Fig. 9). This was due to the different DNA-staining techniques used at MDRS and at the Mesa College Laboratory (see Material and methods section). While at MDRS, precast gels with optimal SYBRsafe concentration for visualization of even lower DNA amounts were used, gels at the Mesa College Laboratory were stained by covering them with ethidium bromide cards. Due to the short incubation time of only 2–4 minutes suggested by the manufacturer (see Material and methods section), the gel might not be evenly penetrated by the ethidium bromide so that the visualization of low amounts of DNA might not be optimal or even below the detection limit. In samples 17–26, no PCR product but the positive control could be detected at MDRS for bacterial and fungal primers (Fig. 7). This was due to a failure during the PCR run at MDRS, based on inconsistent power supply of the habitat generator which happened several times during crew rotations 76 and 77. The signal in the bacterial positive control (Fig. 7, upper row, third gel, lane 9) is likely due to the high starting DNA concentration in the PCR mixture sufficient to generate detectable amounts of amplification product in even low cycle numbers. Samples 27–30, as well as the analysis for archaeal DNA content of the samples, were not investigated at MDRS because of limited time during the simulation campaign. In total, the PCR analysis of soil samples in the MDRS laboratory was highly efficient and comparable to the analysis performed in the Mesa College Laboratory (Fig. 9, Table 3).

Fig. 8. PCR experiments performed at the Mesa College Laboratory. Upper part: PCR with bacterial primers. Middle part: PCR performed with archaeal primers. Lower part: PCR with primers specific for fungi. 01–30: PCR amplification of DNA from collected samples. nc: negative control: reaction with water substituting the DNA. m: commercially available molecular weight marker as a fragment length standard (1 kb Plus DNA ladder; filled arrows indicate the 1.5 kb fragment; open arrows indicate the 0.5 kb fragment). For each PCR sample 25 μl of the reaction mixture was applied to the agarose gel.

Fig. 9. Comparison of PCR experiments performed at MDRS and at the Mesa College Laboratory. Shown are how many of the first 16 samples were positive for bacteria and/or fungi in the PCR experiment at MDRS compared to the PCR run at the Mesa College Laboratory, with the differences in detection level ascribed to different staining techniques sensitivity.
Microbial content with respect to sampling depths and neighbouring vegetation
The total microbial content of all analysed samples is shown in Fig. 10. Out of the 30 samples, 21 contained bacterial DNA (70%), 18 contained fungal DNA (60%), and in 15 samples (50%) archaeal DNA was also detectable. The bacteria were as expected, the predominant micro-organisms, followed by fungi and archaea. This trend was also observed for soil and water samples analysed separately (Fig. 10). Considering the sample depths, it is interesting to see that most of the PCR positive soil samples from depths of 0 to −2 cm contained bacterial DNA (90%), while only 60% of the investigated samples were positive for fungi and 50% were positive for archaea (Fig. 11). At a depth of −10 cm, approximately half of the samples included all 3 types of micro-organisms, while at −30 cm the situation was slightly reversed and the content of archaeal DNA prevailed; however, it would be necessary to analyse more samples to verify the significance of these data. Additionally, the presence of neighbouring vegetation seemed to have an effect on the microbial content of the soil (Fig. 12). In 64–73% of the collected samples, bacterial, fungal or archaeal DNA was found for the samples collected close to a bush, grass or a cactus. At locations without vegetation, microbial DNA was only found in 43–64% of the samples and bacteria were again the domain with the highest occurrence. Nevertheless, it seems that the existence of plants favoured a higher amount of microbial DNA in the soil.

Fig. 10. Total number of samples positive for bacterial, fungal and/or archaeal DNA.

Fig. 11. Influence of the sampling depths on the microbial content. While most of the surface samples (0 to −2 cm) contained bacterial DNA, fungi and archaea prevail at higher depths.
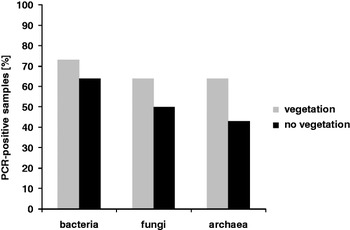
Fig. 12. Influence of neighbouring vegetation on the microbial content of the soil sample.
Discussion and Conclusion
The PCR allows the amplification of the smallest amounts of nucleic acids originating from diverse starting material and is therefore a universal tool for answering questions from very different fields, such as cell and molecular biology, medicine, diagnostics and forensic science. Despite these completely different applications, the PCR process itself is relatively consistent. Surprisingly, although the PCR technique (Saiki et al. Reference Saiki, Scharf, Faloona, Mullis, Horn, Erlich and Arnheim1985) is widely used in many laboratories since its discovery in 1983 by Kary Mullis, it has never before been used as an in-situ microbial DNA detection technique in Mars analogue research stations to test its applicability to the detection of life. In our study, we performed a field experiment in a Mars analogue environment to analyse the possibility of identifying microbial life under the harsh conditions of a simulated exploration mission on the Martian surface, far removed from comfortable, clean and sterile laboratory conditions.
During the preparation for the MDRS mission, we paid attention to the size and ease of handling of all instruments and consumables required for the experiment since limited space would be an issue on a real mission to Mars. Although the machines used for PCR were initially large and heavy, the latest developments have led to battery-operated thermal cyclers of a very small size, the smallest fitting in the palm of a hand (71×121×47 mm; PalmPCR, Ahram Biosystems 2010). Development is still ongoing and we can expect even smaller and lighter instruments in the future. Additionally, a combination of DNA extraction and subsequent PCR analysis in the same automated process would reduce contamination risks and increase the comparability of samples due to a highly standardized procedure. Automated DNA purification instruments capable of isolating minute amounts of DNA for forensic diagnostics already exist, such as the Qiacube (Qiagen 2010) or the Maxwell 16 (Promega 2010). Another advantage would be the integration of a sequencer into this automated process, so that after DNA extraction and PCR amplification with the oligonucleotide primers of choice, the micro-organisms could be immediately identified by their sequence. This combination of automated processes will also be highly interesting for unmanned Mars lander missions and could represent a useful additional tool alongside the Life Marker Chip (LMC), one of the key instruments to detect signs of past and present life on Mars during ESA's ExoMars mission on board the ExoMars rover (Parnell et al. Reference Parnell, Cullen, Sims, Bowden, Cockell, Court, Ehrenfreund, Gaubert, Grant and Parro2007) or during follow-up missions. In contrast to the PCR-based detection of life, the LMC utilizes immunoassays with a fluorescent readout in a microarray format to detect specific organic molecules or classes of molecules (Parnell et al. Reference Parnell, Cullen, Sims, Bowden, Cockell, Court, Ehrenfreund, Gaubert, Grant and Parro2007; Wilson Reference Wilson2007). It would be desirable to develop two independent analytical methods side-by-side to compare, evaluate and confirm the research results.
Ribosomal genes are the most conserved DNA segments, and are present across the kingdoms of life on Earth. Using DNA primers corresponding to the 16S and 18S ribosomal RNA genes, samples from any environment can be analysed for their microbial inhabitants (Isenbarger et al. Reference Isenbarger, Carr, Johnson, Finney, Church, Gilbert, Zuber and Ruvkun2008). Therefore, our focus was to establish the PCR technique for the detection and discrimination of micro-organisms from the three domains (bacteria, archaea and eukarya) using universal primers located in the 16S and 18S ribosomal RNA genes. These ribosomal genes are used frequently for phylogenetic analyses (Woese Reference Woese1987), since they are small enough for laboratory manipulations, but large enough to enable accurate interspecies comparisons. The great advantage of these genes is that certain areas are conserved, while others are highly variable (van de Peer et al. Reference Van de Peer, Chapelle and de Wachter1996) so that PCR experiments can be designed either to identify whole domains or single species, depending upon the chosen primer locations.
Another goal was to analyse the composition of the microbial communities that exist in places with extreme environmental conditions such as the Utah desert. For this reason, samples were collected from different sampling depths at locations both with and without vegetation. The DNA extraction and PCR analysis were performed immediately after sample collection on-site to avoid long-term storage which would most likely falsify the results. Carrying out the data analysis on-site allows the composition of microbial communities to be much more precisely identified, as the DNA is extracted immediately and the micro-organisms are not exposed to abnormal nor hostile conditions.
The analysis of sampling depths (Fig. 11) shows that bacteria, archaea and eukarya are present at each depth with approximately the same frequency, ranging from 44 to 67%, with an exception for the bacterial surface content (at a depth of up to −2 cm). In 90% of the analysed samples, we could identify bacterial DNA. This could be related to the animal excrement or dead plant components serving as nutrients. Due to the strong winds which often prevail in this desert, bacteria are spread throughout the whole area and can be relocated on the soil surface.
We were also interested in the effect of vegetation on the content and composition of microbial communities. All three domains (bacteria, archaea and eukarya) were identified in soils with and without vegetation with a preference for locations with vegetation (Fig. 12). Again, bacteria were the predominant micro-organisms and were detected in 64% of all analysed samples without neighbouring vegetation and in 73% of all samples with vegetation. The increased number of positive samples in cases with neighbouring vegetation could be explained by symbiosis and food supply. Many bacteria such as rhizobia (reviewed in van Rhijn & Vanderleyden Reference Van Rhijn and Vanderleyden1995) and fungi live in symbiosis with plants (Smith & Read Reference Smith and Read1997). Also for archaea of the organotroph nutritional type, the close vicinity of plants is beneficial. Additionally, the soil mineral composition is a crucial point for microbial existence and growth, and is discussed for some of the samples listed in Table 1 in Ehrenfreund et al. (Reference Ehrenfreund, Röling, Thiel, Quinn, Septhon, Stoker, Direito, Kotler, Martins, Orzechowska, Kidd and Foing2011; this volume). A further possibility to investigate the composition of microbial communities on the level of the ribosomal RNA genes is to subclone the amplified PCR products and identify different microbial species by sequencing (Direito et al. Reference Direito, Ehrenfreund, Marees, Staats, Foing and Röling2011; this volume) or even to perform a metagenome sequencing analysis of the entire microbial community (Tringe et al. Reference Tringe, von Mering, Kobayashi, Salamov, Chen, Chang, Podar, Short, Mathur and Detter2005).
In summary, PCR experiments at MDRS demonstrated clearly that despite the prevailing suboptimal, semi-sterile laboratory conditions, it is possible to detect microbial DNA with a quality and sensitivity comparable to a standard molecular biology laboratory. This is an encouraging start and further research should test the identification of micro-organisms by PCR in Mars-like environments to improve the technique and automation of the single steps in the process. Places such as MDRS are indispensable for pre-mission tests to learn as much as possible and be prepared for the problems that could be encountered and how they could be solved.
Apart from the characterization of living microbial communities, the analysis of bacterial DNA signatures is also a suitable method to analyse past microbial communities in sediments (Inagaki et al. Reference Inagaki, Okada, Tsapin and Nealson2005). However, one of the problems associated with the analysis of DNA extracted from sediments is the potential for vertical migration of DNA across strata (Haile et al. Reference Haile, Holdaway, Oliver, Bunce, Gilbert, Nielsen, Munch, Ho, Shapiro and Willerslev2007), which has to be considered and analysed very carefully. Most importantly, surveying for nucleic acid-based life on other planets must be carried out with caution, owing to the risk of contamination by Earth-based life (Poole & Willerslev Reference Poole and Willerslev2007).
In any environment, the flux of nucleic acids due to cell death and horizontal gene transfer is tremendous. After the death of an organism, DNA is rapidly degraded by enzymes or microbial organisms (Eglinton & Logan Reference Eglinton and Logan1991), whereas desiccation or adsorption to a mineral matrix may prevent degradation. Due to enzymatic processes that occur shortly after death and non-enzymatic hydrolytic cleavage of phosphodiester bonds in the phosphate-sugar backbone (Lindahl Reference Lindahl1993; Päabo et al. Reference Päabo, Poinar, Serre, Jaenicke-Despres, Hebler, Rohland, Kuch, Krause, Vigilant and Hofreiter2004) that generate single-stranded nicks, the average size of ancient DNA (aDNA) from Earth is between 100 and 500 bp (Päabo et al. Reference Päabo, Poinar, Serre, Jaenicke-Despres, Hebler, Rohland, Kuch, Krause, Vigilant and Hofreiter2004). As constant low temperatures play a central role in the longevity of aDNA molecules (Lindahl Reference Lindahl1993; Hofreiter et al. Reference Hofreiter, Serre, Poinar, Kuch and Päabo2001; Willerslev & Cooper Reference Willerslev and Cooper2005) and rapid desiccation and high-salt concentrations are also strong DNA survival promoting factors (Lindahl Reference Lindahl1993), the Mars environment appears very well suited for long-time survival of DNA. Additionally, nucleic acid functional groups, including sugar hydroxyl groups, phosphate groups and extracyclic functional groups on the bases, are capable of binding to minerals (Cleaves et al. Reference Cleaves, Jonsson, Jonsson, Sverjensky and Hazen2010) and, if attached to a surface, are protected and survive strong UV exposure (Lyon et al. Reference Lyon, Monier, Dupraz, Freissinet, Simonet and Vogel2010).
The space environment with vacuum, extreme dehydration and solar and galactic cosmic radiation, prevents the survival of most organisms in space (Horneck Reference Horneck, Rothschild and Lister2003). Despite this extremely challenging situation for life, there are organisms on Earth which are potentially able to survive space travel: it has been shown that different micro-organisms can survive launch by spallation from a hypervelocity impact (Horneck et al. Reference Horneck, Stöffler, Ott, Hornemann, Cockell, Moeller, Meyer, de Vera, Fritz and Schade2008a; Fajardo-Cavazos et al. Reference Fajardo-Cavazos, Langenhorst, Melosh and Nicholson2009) and hypervelocity atmospheric transit (Fajardo-Cavazos et al. Reference Fajardo-Cavazos, Link, Melosh and Nicholson2005). Thus, it has been suggested that, for example, bacterial spores situated on or within meteorites could survive interplanetary transport (Fajardo-Cavazos et al. Reference Fajardo-Cavazos, Langenhorst, Melosh and Nicholson2009) and hypervelocity entry from space through Earth's atmosphere (Fajardo-Cavazos et al. Reference Fajardo-Cavazos, Link, Melosh and Nicholson2005; reviewed in Olsson-Francis & Cockell Reference Olsson-Francis and Cockell2010). However, DNA may be the sensitive target of spores exposed to ultrahigh shock pressures (Moeller et al. Reference Moeller, Horneck, Rabbow, Reitz, Meyer, Hornemann and Stöffler2008). As another extreme example, tardigrades, which are among the most desiccation and radiation-tolerant animals, have been shown to survive extreme levels of ionizing radiation (Horikawa et al. Reference Horikawa, Sakashita, Katagiri, Watanabe, Kikawada, Nakahara, Hamada, Wada, Funayama and Higashi2006), temperatures of −273°C (Becquerel Reference Becquerel1950) and the vacuum of space (Jönsson et al. Reference Jönsson, Rabbow, Schill, Harms-Ringdahl and Rettberg2008). Whereas microgravity and radiation had no effect on the survival nor DNA integrity of active tardigrades (Rebecchi et al. Reference Rebecchi, Altiero, Guidetti, Cesari, Bertolani, Negroni and Rizzo2009), after being exposed to the space environment for 12 days, tardigrades molted and females laid eggs (Rebecchi et al. Reference Rebecchi, Altiero, Guidetti, Cesari, Bertolani, Negroni and Rizzo2009). Several eggs hatched, and the newborns exhibited normal morphology and behaviour (Rebecchi et al. Reference Rebecchi, Altiero, Guidetti, Cesari, Bertolani, Negroni and Rizzo2009).
Therefore, interplanetary transport of DNA or even of living organisms from Mars to Earth, especially in the early phase of solar system development, is neither impossible nor unlikely, but a possible hypothesis. If life on Mars is not based on DNA, it would support the hypothesis that life developed independently from Earth and that the special conditions on Mars also allowed development of life in another form than that on Earth, strongly suggesting that life could be a likely event in a reasonably wide range of planetary conditions. But if life on Mars is based on DNA, there is the possibility that material was transferred between planets. The results from different Mars orbiters and landers suggest environmental conditions allowing potential microbial growth: areas with subsurface water and sulphur, as well as rocks for endoliths and permafrost regions, all of which are of interest as potential sites of existing life (Horneck Reference Horneck2008b). In a recent study simulating Martian atmospheric conditions and subsurface water in the form of ice, it was shown that the growth of non-extremophile bacteria was possible (Pavlov et al. Reference Pavlov, Shelegedina, Vdovina and Pavlov2010) indicating that there are hypothetical habitats on Mars where life could still exist. In the following decades, there will be a chance to address this eminent question about the existence of life on Mars and its similarity to terrestrial life by sample return, robotic or manned missions to Mars and modern PCR-based analysis techniques.
Acknowledgements
The EuroGeoMars 2009 campaign was organized and supported by the International Lunar Exploration Working Group (ILEWG), NASA Ames Research Center and ESA/ESTEC. We acknowledge the contribution of the EuroGeoMars 2009 campaign crew and the mission support team. We thank in particular Carol Stoker (NASA Ames Research Center) and Jason Page (ESTEC) for experimental support. C.S.T. acknowledges travel support to MDRS from a grant from ILEWG. The help of Denise McKenny (Mesa State College, Grand Junction, Colorado), Jörn Kalinowski (University of Bielefeld), Heather Smith and Chris McKay (NASA Ames Research Center), was also highly appreciated for collaborations and fruitful discussions. Furthermore, we thank Uwe Schlomann, Garrit Koller, Tina Warinner, Sara Duke and John B. McClean for critically reading the manuscript and providing constructive suggestions.















