


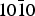

Introduction
Life on Earth, at least as we know it today, is inseparable from homochirality. Therefore, if we consider homochirality as a prerequisite for the emergence of life, the presence of an enantiomeric excess in the organic matter of well-defined families of meteorites (Cronin & Pizzarello Reference Cronin and Pizzarello1997; Pizzarello & Cronin Reference Pizzarello and Cronin1998, Reference Pizzarello and Cronin2000) is a strong argument in favour of the Panspermia Hypothesis proposed by Richter circa 1865, which claimed that the seeds of life are present all over the Universe.
However, not only is the origin of the enantiomeric excess unknown, whatever the place it is supposed to have appeared, but the emergence itself of the first chiral system is still an unanswered question. Not even a single chiral molecule has been identified so far in the interstellar medium (ISM) that could provide a clue to the mechanism at work in their formation. Nevertheless, the complexity and population of the interstellar observed molecules, known under the generic name of complex organic molecules (COMs), are growing day after day, thanks to the improvements of the observational techniques. As an encouraging example, it can be quoted that the first ramified species, isopropylcyanide (CH3CHCH3-CN) has just been discovered in the ISM (Belloche et al. Reference Belloche, Garrod, Müller and Menten2014). There is no rational argument to be developed not to detect a chiral molecule soon. We proposed recently (Marloie et al. Reference Marloie, Lattelais, Pauzat and Ellinger2010) a number of chiral candidates for observation that are at the same time the most stable isomers that can be formed (Lattelais et al. Reference Lattelais, Pauzat, Ellinger and Ceccarelli2009) from a given set of atoms (among which lactic acid that will be considered below).
Within the hypothesis that an enantiomeric choice cannot be made ex nihilo, i.e. without the participation of an external agent capable of inducing homochirality, we focused on possible means of selecting one enantiomer over the other. Among the different hypotheses suggested in the literature, we concentrate on the selective adsorption mechanism. Two enantiomers, indeed, have identical internal energies but, when adsorbed on a same chiral surface, the complexes obtained are no longer enantiomers but diastereoisomers. These diastereoisomers do not have the same energy so that one should be more strongly attached to the surface than the other. Assuming the pre-existence of a chiral surface, we address the question of whether the adsorption energy differs significantly enough from one enantiomer to the other to create a difference between their populations on the surface of the solid phase when starting from a racemic mixture. The present study being restricted to an interstellar context, we considered only solid surfaces and gas phase molecules, ruling out any intervention of a liquid phase as a solvent.
Though irrelevant to the interstellar context, it has to be reminded that enantio-specific adsorption has been extensively considered on metal surfaces (Bhatia & Sholl Reference Bhatia and Sholl2005; Gladys et al. Reference Gladys, Stevens, Scott, Jones, Batchelor and Held2007; Held & Gladys Reference Held and Gladys2008) in the frame of periodic density functional theory (DFT)Footnote 1. The chiral Cu surface considered in the calculations being also available for experiments, temperature programmed desorption studies (TPD) were performed on these surfaces (Horvath & Gellman Reference Horvath and Gellman2001, Reference Horvath and Gellman2002; Horvath et al. Reference Horvath, Koritnik, Kamakoti, Sholl and Gellman2004) and allowed useful comparison validating the computational approach.
Some natural materials, such as kaolin, are also chiral, and a theoretical study done about 25 years ago (Julg Reference Julg1989) already suggested that enantio-specific adsorption of amino acids was indeed possible at their surfaces. The experimental problem with kaolin is that it has not yet been possible to dispose of chiral single crystals. Adsorption on another natural chiral surface, namely that of calcite, has been studied more recently (Asthagiri & Hazen Reference Asthagiri and Hazen2007) on the example of alanine. Results were deceitful but, as stressed by the authors themselves, the dimensions of the substrate unit cell were probably not well adapted to that of the adsorbate molecule. Then α-quartz, i.e. chiral due to the L or D arrangement of the SiO2 units in helicoidally structures was also considered as solid support for adsorption by Han & Sholl (Reference Han and Sholl2009, Reference Han and Sholl2010). Performing plane wave DFT calculations, the authors studied the enantiomers of several amino acids and obtained mixed results. Some values are barely significant, for example those concerning alanine, which raises the question of the relevance of both the theoretical approach (as mentioned by the authors) and that of the chiral surface considered (Downs & Hazen Reference Downs and Hazen2004). All these investigations, whatever the results obtained in the end, revealed that the selectivity is strongly depending of the site chosen on the surface; the complexity of the surfaces like those of α-quartz could even generate adsorption sites with opposite selectivity for the same chiral molecule. In such conditions, it is clear that local adsorption could not be representative of the real selectivity of a surface and might lead to erroneous conclusions. The results of extensive computational as well as experimental studies, carried out on the adsorption of biomolecules on achiral silica surfaces can be found in the review by Rimola et al. (Reference Rimola, Costa, Sodupe, Lambert and Ugliengo2013); the computational studies reported there were also done using DFT formalism, but in a different way since they used a finite cluster approach instead of an infinite surface.
In this paper, working with a periodic description of the infinite surface, we proposed a different approach, namely that the selectivity of the surface be considered as a global property and determined by a statistical treatment.
Models
The dia-stereo complexes formed by adsorption of the two enantiomers have different energies contrary to the free-flyer species. This property induces the possibility to adsorb one of the two enantiomers selectively, and thus to engage a process of enantiomeric enrichment on the solid support. In this context, the purpose of the study is to evaluate how different are the responses of the two enantiomers to the adsorption phenomenon by determining the difference between their adsorption energies on the same chiral surface. The procedure is illustrated below on the two examples of α-alanine and lactic acid. These target molecules (Fig. 1) were chosen for the following reasons:
(i) α-Alanine, H2N-CH(CH3)-COOH is the simplest chiral amino acid. It is present in living organisms and also abundant in meteorites (see, e.g., Martins & Sephton Reference Martins, Sephton and Hughes2009). It has been widely studied for these reasons though showing poor sticking properties, unfortunately (Rimola et al. Reference Rimola, Sodupe and Ugliengo2009),
(ii) Lactic acid, HO-CH(CH3)-COOH is the simplest chiral hydroxy-acid where –OH replaces –NH2 in alanine. In addition, it is one of the best targets for the detection of a chiral molecule in the ISM as it is the most stable isomer of C3H6O3 formula according to the minimum energy principle (Lattelais et al. Reference Lattelais, Pauzat, Ellinger and Ceccarelli2009) and has a rather large dipole moment with a value of 2.3 Debye (Marloie et al. Reference Marloie, Lattelais, Pauzat and Ellinger2010) favourable to its detection.

Fig. 1. Schematic representations of R-alanine (left) and R-lactic acid (right) enantiomers
As solid support, we chose silica whose presence in the ISM has been disputed for a long time but that has been identified recently in the envelope of T-Tauri stars (Sargent et al. Reference Sargent2009). Moreover, natural single crystals can be obtained easily in view of laboratory experiments. This combination of adsorbate and substrate has already been considered using a model surface of hydroxylated quartz (Han & Sholl Reference Han and Sholl2009). Due to the abundance of hydrogen in space, the grain surfaces are indeed supposed to be hydroxylated, which means that there are no free oxygen atoms left on the surface. This assumption, relying on astrophysical grounds, is well in line with the DFT calculations by Goumans et al. (Reference Goumans, Wander, Brown and Catlow2007) that showed that the hydroxylated form of the α-quartz {0001} surface was more stable than the raw surface obtained by the simple cut of a single crystal. In this study, we kept the same hydroxylated mineral, namely α-quartz but chose to look at the possibilities opened by the {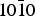 $10\bar 10$} surface supposed to be more responsive to adsorption according to Downs & Hazen (Reference Downs and Hazen2004).
$10\bar 10$} surface supposed to be more responsive to adsorption according to Downs & Hazen (Reference Downs and Hazen2004).
Computational methods
We rely on numerical simulations based on a periodic description of the solid substrate by means of DFT. A plane-wave expansion of the basis set coupled to projector augmented waves (PAW) ultrasoft pseudo-potentials was employed, associated with the PW91 functional (Perdew & Wang Reference Perdew, Ziesche and Eschrig1991), as implemented in the VASP simulation package (Kresse & Hafner Reference Kresse and Hafner1993, Reference Kresse and Hafner1994). The calculations were performed using a 12 × 12 × 1 Monkhorst Pack generated grid for the basic unit cell, with a kinetic energy cut-off at 500 eV; the grid was adapted to provide an equivalent treatment for all the unit cells considered. More computational details are given in Lattelais et al. (Reference Lattelais2011) and Pauzat et al. (Reference Pauzat, Lattelais, Ellinger and Minot2011); for the interested reader, see Wesolowski et al. (Reference Wesolowski, Parisel, Ellinger and Weber1997, Reference Wesolowski, Ellinger and Weber1998); an extensive review of the mathematical properties of the PW91/PW91 functional compared with other GGA functionals and exact results can be found in Filippi et al. (Reference Filippi, Gonze, Umrigar and Seminario1996).
Calculation protocols
Taking into account that periodic calculations are based on the replication of the unit cell along the three directions of space, the size of the unit cell is a critical parameter that has to be determined in order to avoid any spurious interactions. After careful calibration aimed at insuring a ±0.004 eV (~0.1 kcal mol−1) stability of the calculated energiesFootnote 2 (see Appendix A) we used a unit cell with a reactive surface of (9.826 × 10.81) Å2, a vertical dimension of 25 Å, and a slab thickness of 7.1 Å for all the calculations. Briefly, the calculation of a single adsorption site unfolds as follows:
(i) optimizing the geometry of the active surface, keeping the rest of the solid frozen, and calculating the energy, E substrate, of the surface of the substrate entity alone;
(ii) optimizing the geometry of the adsorbate, alone in the cell, and calculating the corresponding energy E adsorbate;
(iii) optimizing the geometry of the adsorbate/substrate complex. The optimization takes into account only the molecule and the adsorption site, i.e. the atoms of the surface interacting directly with the adsorbed molecule (the rest being kept frozen), and delivers the total energy E;
(iv) calculating the adsorption energy E ads as
 $${E_{{\rm ads}}} = ({E_{{\rm substrate}}} + {E_{{\rm adsorbate}}}) - E,$$
$${E_{{\rm ads}}} = ({E_{{\rm substrate}}} + {E_{{\rm adsorbate}}}) - E,$$
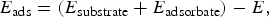
where E is the total energy of the substrate/adsorbate complex.
Based on this protocol, a series of calculations showed that several sites of adsorption can be found on the same surface with different adsorption energies and even different selectivities (see the next section Fig. 2). Such results question the validity of the local answer to molecular adsorption. This led us to consider a systematic search for all possible sites and to use a statistical approach so to deduce an average response of the surface to the adsorption process. The procedure consists of five successive stages detailed in Appendix B:
(i) Building a grid of points on top of the surface to initiate the local (and only local) optimization of the adsorbate/substrate complex, which gives 630 initial guesses for alanine and 1680 for lactic acid.
(ii) Optimizing these different sites (molecule + adsorption points on the surface), by means of the semi-empirical Density Functional based Tight Binding (DFTB+) method (Seifert et al. Reference Seifert, Porezag and Frauenheim1996; Elstner et al. Reference Elstner, Porezag, Jungnickel, Elsner, Haugk, Frauenheim, Suhai and Seifert1998), which leads fast to n relevant points: ~500 for alanine and ~1500 for lactic acid (see Appendix B for details on the procedure).
(iii) Sorting out groups of k points, considered as identical within periodic constraints, on the basis of geometric criteria. A statistical weight of k/n which is its occurrence number normalized to 100% is affected to each group.
(iv) Performing a complete treatment of the representatives (both enantiomers) of each selected group with the usual ab initio first-principle DFT.
(v) Calculating the adsorption energy that is characteristic of the couple adsorbate/surface as the sum of the R (respectively S) adsorption energies of the most significant sites weighted by their occurrence numbers normalized to 100%.

Fig. 2. Adsorption energies and selectivity of R- and S-alanine enantiomers for five selected positions on the {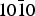 $10\bar 10$} surface of α-quartz. Colour code: grey, R-alanine; white, S-alanine; black, R versus S adsorption selectivity.
$10\bar 10$} surface of α-quartz. Colour code: grey, R-alanine; white, S-alanine; black, R versus S adsorption selectivity.
Results
Adsorption as a local property
Searching for the deepest adsorption well (corresponding to the largest adsorption energy) of alanine, we selected at random five positions among those thought to be the most favoured adsorption sites according to ‘chemical intuition’. All of them were verified to be stable minima but the adsorption energies fluctuate by ±5 kcal mol−1 around a median value ~15 kcal mol−1 (see Fig. 2).
The difference between the adsorption energies of the R- and S-alanine enantiomers, i.e. the R versus S adsorption selectivity, was found to vary over a range of 2 kcal mol−1. Three points out of five give R-alanine more stable than S-alanine, but only one of them presents a significant selectivity above 1 kcal mol−1; one shows no selectivity and one gives a reverse selectivity with S- more stable than R-alanine.
Intuitively, alanine was first positioned so as to present the largest possible tripod in view of providing, supposedly, a strong triple binding (adsorption site 1).
The optimization of the structure showed, indeed, three points of attachment maintaining alanine on the surface (see Fig. 3). The oxygen of the carboxyl group is interacting with a dandling hydrogen (1.64 Å), while the acidic hydrogen of the HO–C=O group is attached to an oxygen atom of the substrate surface (1.78 Å). The third link to the surface is by means of the amino group; the bonding is effective, though weaker, by means of the interaction of one of the NH bonds with an oxygen of the substrate (2.67 Å). In this situation, alanine makes a bridge between two crests over a valley of the substrate surface. The adsorption energy is ~18 kcal mol−1. However, another adsorption site (5) leads to a stronger adsorption energy ~22 kcal mol−1. In this situation, we found also three points of attachment to maintain alanine on the surface (see Fig. 4). Here, the oxygen of the carboxyl group is interacting with a dandling hydrogen (1.98 Å), while the acidic hydrogen of the HO–C=O group is strongly attached to an oxygen atom of the substrate surface (1.70 Å). The third link to the surface is by means of the amino group; but, contrary to site 1, the bonding is effective by means of the N lone pair interacting with a dandling OH bond of the substrate; it is the shortest/strongest hydrogen bond (1.65 Å) that makes the difference between the adsorption sites. In this situation, where the CH3 substituent is as far away from the surface as possible, alanine makes a bridge over a valley between two crests of the substrate surface.

Fig. 3. Adsorption of R-alanine on the { $10\bar 10$} surface of α-quartz for intuitively selected position (1). Colour code: Si, yellow; O, red; N, blue; C, brown; H, white.
$10\bar 10$} surface of α-quartz for intuitively selected position (1). Colour code: Si, yellow; O, red; N, blue; C, brown; H, white.

Fig. 4. Adsorption of R-alanine on the { $10\bar 10$} surface of α-quartz for the most favoured position (5). Colour code: Si, yellow; O, red; N, blue; C, brown; H, white.
$10\bar 10$} surface of α-quartz for the most favoured position (5). Colour code: Si, yellow; O, red; N, blue; C, brown; H, white.
Adsorption as a global property: R- versus S-alanine
In these conditions it seems clear that looking for the adsorption minimum by a priori selection is highly hazardous. A statistical approach taking the whole surface into account looks more appropriate.
When applied to alanine, the five-step approach shows six relevant groups with significant (≥5%) populations, representing 66% of the initial set of sites. Only three of these representative structures correspond to one of those initially considered following chemical intuition. The missing structures correspond to under-populated sites, which have been eliminated for that reason. In particular adsorption site (1) is not part of the statistically significant groups, which confirms the necessity for a systematic screening. It is grateful that site (5) is at the same time the most frequent and the most effective
Taking into account the adsorption energies of the most significant groups, as presented in Fig. 5, and calculating the average selective adsorption energy, we obtain average adsorption energies of 19.0 kcal mol−1 (0.824 eV) and 17.8 kcal mol−1 (0.772 eV) for (R)- and (S)-alanine, respectively. In the end, the statistical budget for alanine on the { $10\bar 10$} surface of α-quartz shows a differential adsorption favouring the R enantiomer by 1.2 kcal mol−1 (0.052 eV).
$10\bar 10$} surface of α-quartz shows a differential adsorption favouring the R enantiomer by 1.2 kcal mol−1 (0.052 eV).

Fig. 5. Statistical budget for alanine adsorption on the { $10\bar 10$} surface of α-quartz. All adsorption energies are in kcal mol−1. Colour code: grey, R-alanine; white, S-alanine; black, R versus S adsorption selectivity
$10\bar 10$} surface of α-quartz. All adsorption energies are in kcal mol−1. Colour code: grey, R-alanine; white, S-alanine; black, R versus S adsorption selectivity
Adsorption as a global property: R- versus S-lactic acid
Applying the same protocol to the case of the lactic acid, we found 15 relevant groups out of which we kept the six with a population ≥5% which represent 67% of the total population (Fig. 6).

Fig. 6. Statistical budget for lactic acid adsorption on the {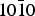 $10\bar 10$} surface of α-quartz. All adsorption energies are in kcal mol−1. Colour code: grey, R-lactic acid; white, S-lactic acid; black, R versus S adsorption selectivity.
$10\bar 10$} surface of α-quartz. All adsorption energies are in kcal mol−1. Colour code: grey, R-lactic acid; white, S-lactic acid; black, R versus S adsorption selectivity.
Average adsorption energies are close whatever the absolute configuration of the enantiomers, namely, 13.1 kcal mol−1 (0.568 eV) and 13.8 kcal mol−1 (0.598 eV) for the R- and S-lactic acid, respectively. Four, out of the six groups selected, favour the R enantiomer, but with a marginal selectivity ~0.3 kcal mol−1. The global selectivity of −0.7 kcal mol−1 (~−0.03 eV) comes essentially from two groups representing ~25% of the adsorbed population. Finally, the statistical budget for lactic acid on the α-quartz {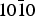 $10\bar 10$} surface shows a differential adsorption favouring the S enantiomer by <1 kcal mol−1. The most favoured position representing 9.3 % of the adsorbed population is represented below (Fig. 7).
$10\bar 10$} surface shows a differential adsorption favouring the S enantiomer by <1 kcal mol−1. The most favoured position representing 9.3 % of the adsorbed population is represented below (Fig. 7).

Fig. 7. Adsorption of S-lactic acid on the 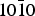 $10\bar 10$ surface of α-quartz for the most favoured position. Colour code: Si, yellow; O, red; C, brown; H, white.
$10\bar 10$ surface of α-quartz for the most favoured position. Colour code: Si, yellow; O, red; C, brown; H, white.
This most favoured adsorption site presents a local R selectivity, opposite to that obtained from the statistical S preference for the whole surface. Its position on the surface is consistent with that of R-alanine on site (1). Both species have similar adsorption energies, both with two strong hydrogen bonds, one between the carboxyl oxygen and a dandling OH of the substrate, the other between the acidic hydrogen and an oxygen on a crest on the substrate surface, together with the CH3 group parallel to the surface.
Discussion
As shown above, several sites of adsorption can be found on the same surface with different adsorption energies and even a different selectivity. It is not really surprising in view of the complexity of the surface with alternating valleys and ridges. Deciding of adsorption sites on chemical intuition only, shows clearly its limitations. It should be kept in mind that strong adsorption situations may not be statistically significant because of insufficient population (alanine site 1), but also that the highest adsorption situation may not be discriminating correctly between enantiomers (lactic acid).
Considering the substrate surface as a whole, with all the possible adsorption sites, seems a better approach. At temperatures in the range of 10 to 50 K, the thermal energy contribution is negligible. Medium size organic species with masses large enough to accommodate a chiral centre with a carboxylic function are not really inclined to hopping on the surface. With adsorption energies between 12 and 20 kcal mol−1 (i.e. between 6000 and 10 000 K), they will stick to the surface with a probability proportional to the number of adsorption sites.
Without the availability of a thermal bath to borrow external energy, we chose to determine the adsorption sites and adsorption energies starting from initial conditions covering all possibilities of conformations for the adsorbates over the substrate surface. The square grid-pattern with a 0.4 Å step, i.e. one-tenth of the extension of the adsorbed molecules defines a mesh of initial guesses that is dense enough to meet that purpose. In spite of the high number of structures to consider, this first screening was made possible using DFT in the tight binding approximation (DFTB+ method). This semi-empirical method, ~400 times faster than first principle DFT, is well adapted to locate the structural characteristics of the molecule/surface complexes. The grouping of more than ~500 (alanine) and ~1500 (lactic acid) points according to their geometric similarity led to a reduced number of significant families. This systematic search starting from a high-density grid of point makes possible eliminating a great number of geometries that should have been considered otherwise in first-principle calculations. However, this semi-empirical approach is not able to provide the desired precision for absolute energies. It is the reason why the structures and adsorption energies had to be recalculated for every leading group by means of first-principle periodic DFT.
In the end the results obtained for the adsorption of α-alanine and lactic acid on the α-quartz {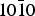 $10\bar 10$} surface show that the (R) enantiomer of alanine is preferentially adsorbed by 1.2 kcal mol−1 (~0.05 eV), while the (S) enantiomer of lactic acid is preferentially adsorbed by 0.7 kcal mol−1 (~0.03 eV) on the same surface. The results of our statistical approach confirm those of a preceding study for alanine in which the α-quartz surfaces considered were found poorly enantio-selective. The {
$10\bar 10$} surface show that the (R) enantiomer of alanine is preferentially adsorbed by 1.2 kcal mol−1 (~0.05 eV), while the (S) enantiomer of lactic acid is preferentially adsorbed by 0.7 kcal mol−1 (~0.03 eV) on the same surface. The results of our statistical approach confirm those of a preceding study for alanine in which the α-quartz surfaces considered were found poorly enantio-selective. The {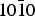 $10\bar 10$} surface is not doing any better for lactic acid. The poor selectivity obtained in both cases may be attributed to the similar behaviour of the adsorption sites, all found with three hydrogen bonds hooked to the surface for the two enantiomers.
$10\bar 10$} surface is not doing any better for lactic acid. The poor selectivity obtained in both cases may be attributed to the similar behaviour of the adsorption sites, all found with three hydrogen bonds hooked to the surface for the two enantiomers.
Conclusion
The quartz {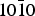 $10\bar 10$} surface is found capable of trapping chiral species bearing two different functional groups (here alanine and lactic acid) with significant adsorption energies. However, these energies can vary on the same surface according the site from 0.5 to 1 eV. Furthermore, and whatever the adsorption site, the selectivity, i.e. the energy difference between the two enantiomers, is weak and may even change sign. Considering the surface as a whole is then the most reasonable attitude.
$10\bar 10$} surface is found capable of trapping chiral species bearing two different functional groups (here alanine and lactic acid) with significant adsorption energies. However, these energies can vary on the same surface according the site from 0.5 to 1 eV. Furthermore, and whatever the adsorption site, the selectivity, i.e. the energy difference between the two enantiomers, is weak and may even change sign. Considering the surface as a whole is then the most reasonable attitude.
After statistical averaging of the adsorption energies, we come to the fact that the quartz {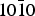 $10\bar 10$} surface is poorly selective, in contradiction to what was expected from the chiral index study for mineral surfaces based on the crystal symmetry (Downs & Hazen Reference Downs and Hazen2004). However, making a general statement about selectivity, given the specific surface and chiral systems used, may be hazardous for at least two reasons. On the one hand, the chiral molecules considered, H2N-CH(CH3)-COOH and HO-CH(CH3)-COOH, have the functional groups responsible of the adsorption both on the same carbon; it might be different for larger systems where remote functional groups could reach different adsorption sites from one enantiomer to the other. On the other hand, natural surfaces are not perfect but present defects such as steps, adatoms, kinks, etc. that could be able to generate stronger interactions and above all, interactions more specific of each enantiomer.
$10\bar 10$} surface is poorly selective, in contradiction to what was expected from the chiral index study for mineral surfaces based on the crystal symmetry (Downs & Hazen Reference Downs and Hazen2004). However, making a general statement about selectivity, given the specific surface and chiral systems used, may be hazardous for at least two reasons. On the one hand, the chiral molecules considered, H2N-CH(CH3)-COOH and HO-CH(CH3)-COOH, have the functional groups responsible of the adsorption both on the same carbon; it might be different for larger systems where remote functional groups could reach different adsorption sites from one enantiomer to the other. On the other hand, natural surfaces are not perfect but present defects such as steps, adatoms, kinks, etc. that could be able to generate stronger interactions and above all, interactions more specific of each enantiomer.
A point to be emphasized in this study is the use of the statistical treatment that brings a challenging perspective for interpreting the selectivity of adsorption processes, especially when small energy differences are involved, which is the case for enantiomeric imbalance. It does not seem necessary when are implied large energy differences such as those found in a preceding study of the differential adsorption between isomers on crystalline ice (Lattelais et al. Reference Lattelais2011). The relevance of a statistical treatment can be questioned according to the complexity and coverage of the surface. In case of low coverage, the energetically most favoured position could be the only one to consider if diffusion at the surface is important or if the radiation flux is sufficient to induce non-destructive photo-desorption allowing re-adsorption on opportune positions. For a coverage close to a monolayer, all sites will most probably be occupied and a statistical behaviour should be observed. In the case of several layers of adsorbed molecules, the molecule/molecule interaction may be dominant and the solid surface underneath is not seen; the present considerations are not relevant in such cases.
At last, from a computational point of view, it has to be stressed that it is the combination of both, the semi-empirical DFTB+ method for the largest possible screening of the adsorption geometries and the first principle periodic DFT method for the fine tuning of the structures and adsorption energies of the adsorbate/substrate complexes, which provides a powerful and economic tool for such a search.
Acknowledgements
This research was supported by grants from CNRS programmes EPOV (Planetary Environments and Origins of Life) and PCMI (Physique et Chimie du Milieu Interstellaire). Parts of the numerical simulations were performed using HPC resources from GENCI-CINES (Grant no. 2012-085128).
Appendix A
The basal and the vertical dimensions (as shown in Fig. A1) have been checked according to the procedure presented below.
(i) The slab used for modelling the {
$10\bar 10$} surface is obtained by an appropriate cut of the top three silicon layers of the experimental crystal structure. The reactive surface dandling bonds of the oxygen atoms are saturated with hydrogen atoms whose positions are optimized. The silicon atoms on the bottom (non-reactive surface) are saturated by hydrogen atoms positioned in the direction of the missing oxygens at a distance SiH de 1.5 Å. To be safe from any unwanted interaction between the silica sheets generated by the 3D periodic treatment, a height of 25 Å high was taken for the unit cell (corresponding to a vacuum of 18 Å). It is a conservative value, arbitrarily chosen, in view of previous adsorption studies (Pauzat et al. Reference Pauzat, Lattelais, Ellinger and Minot2011; Lattelais et al. Reference Lattelais2011). The surface of the α-quartz minimum unit cell (4.913 × 5.405) Å2 is not large enough to allow the adsorption of even the smallest amino acid, glycine (i.e. 4.3 Å long in its stable conformation) without facing important lateral interactions between nearest neighbours. In order to remove these lateral interactions, i.e. those corresponding to approaching isolated species, we have determined the minimum surface beyond which the energy of the adsorbate (here α-alanine) does not vary when the dimensions are increased. The energy of the molecule, alone in the unit cell, was then calculated for increasing values of the horizontal surface area, which is equivalent to probing the energy of the molecule as a function of its separation from its nearest neighbours.From the results in Table A1 it can be seen that increasing the surface by a factor of four with respect to the (10 × 10) Å2 area, taken arbitrarily as reference, leads only to an extra stabilization of 5.7 10−2 kcal mol−1, which appears negligible. As a consequence, we used a (9.826 × 10.81) Å2 surface for the unit cell in all adsorption calculations, i.e. four times the size of the α-quartz minimum unit cell.
(ii) All possibilities of vertical interactions arising from the fact that the surface replicate above the adsorbed molecule might possibly interact with the molecule have to be removed. Using the surface defined above, the adsorption energy of α-alanine was calculated as a function of the vertical dimension of the cell. We found that a vacuum of 13–23 Å between two consecutive slabs does not change the calculated adsorption energy within the desired threshold of ±0.05 kcal mol−1 (Table A2).
(iii) The last parameter to be checked is the thickness of the slab to be sure that the bulk is large enough to prevent any possible bias coming from an interaction of the bottom surface with the adsorbed molecule through the slab itself. Adding a fourth layer of Si atoms, we found that the adsorption energy of (R)-alanine is marginally modified. More precisely, an increase of the thickness of the slab from 7.1 to 11.5 Å results in a decrease of the adsorption energy from 18.6 to 18.5 kcal mol−1, that is ~1%, which can be considered negligible.


Fig. A1. Calibration of the basal (left) and vertical (right) dimensions of the unit cell.
Table A1. Decrease of the lateral interaction energies ∆E (kcal mol−1) arising from replication as a function of the basal surface (Å2) of the unit cell

Table A2. Adsorption energies E ads (kcal mol−1) of R-alanine as a function of the height (Å) of the unit cell

Appendix B
To avoid the obvious drawbacks of the trial and error method in the search for the hypothetical minimum of a complex molecule interaction with a complex surface, we used an a priori procedure organized in five successive steps.
(i) The first stage is the building of a grid of points on top of the surface at a distance of 2.5 Å. The distance was chosen so that the initial interaction with the surface is effective enough to activate the local (and only local) optimization of the adsorbate/substrate complex. The grid has a square pattern with a 0.4 Å regular step, which amounts to 210 different points for the minimum unit cell. Multiple orientations of the adsorbed molecule were considered for each point of the grid (three for alanine or eight for lactic acid), which gives 630 initial guesses for alanine and 1680 for lactic acid. We found it necessary after observing that different starting orientations could lead to different optimized geometries and adsorption energies.
(ii) The second stage consists in optimizing these different sites (molecule + adsorption points on the surface), using the usual extended unit cell. For that purpose we employed the semi-empirical DFTB+ method (Seifert et al. Reference Seifert, Porezag and Frauenheim1996; Elstner et al. Reference Elstner, Porezag, Jungnickel, Elsner, Haugk, Frauenheim, Suhai and Seifert1998) which is about 400 times faster than first-principle DFT and consequently allows the screening of such a number of points in a reasonable computing time. This procedure is completed by the elimination of the points considered irrelevant as: unbounded molecules, non-converged geometries, orphan structures, which finally leads to n relevant points: ~500 for alanine and ~1500 for lactic acid.
(iii) The third stage aims at regrouping the adsorption sites obtained in the preceding step by geometrical similarity, in order to define typical adsorption structures. Thus, groups of k points, considered as identical within periodic constraints, were sorted out on the basis of geometric criteria using an automated procedure. A statistical weight of k/n is affected to each group, i.e. its occurrence number normalized to 100%. For alanine, nine relevant groups were found, whose most representative one (~20%) corresponds to the point #5 (see Fig. 2) previously found by a trial and error search based on chemist's intuition. In the case of lactic acid, 15 groups were selected: two with a population <1%, seven with a population between 1 and 5% and six with a population over 5% representing ~70% of the total population.
(iv) The fourth stage considers the representatives (both enantiomers) of each selected group and performs a complete optimization treatment of the structure with the usual first-principle periodic DFT.
(v) Finally the adsorption energy that is characteristic of the couple adsorbate/surface is obtained as the sum of the R (respectively S) adsorption energies of the most significant sites weighted by their occurrence numbers normalized to 100%.










