


Introduction
Phosphorus is the 18th most abundant element in the cosmos, 11th most abundant within our planet and, alongside C, H, O, N and S, is a central element in terrestrial biochemistry (Macia Reference Macia2005). On Earth, phosphorus occurs naturally as fully oxidized orthophosphate (PO43−) most commonly within igneous and metamorphic rocks of the apatite family Ca5(PO4)3(X) (X=Cl, F, OH), wherein it is estimated that some 8×1017 kg of phosphate exists on land and within ocean beds (Van Cappellen & Ingall Reference Van Cappellen and Ingall1996). Nature has evolved sophisticated processes to exploit orthophosphates in terrestrial biochemistry (Lodish et al. Reference Lodish, Berk, Zipursky, Matsudaira, Baltimore and Darnell2000), based almost entirely upon non-redox processes involving catalysed hydrolysis of the phosphorus–oxygen (P–O) bond (Westheimer Reference Westheimer1987). However, Gulick (Reference Gulick1955) identified problems with the use of orthophosphate in the earliest stages of prebiotic chemistry, chiefly poor water solubility of metal salts and low inherent chemical reactivity in the absence of activating chemicals (Österberg Reference Österberg, Orgel and Lohrmann1973; Leman et al. Reference Leman, Orgel and Ghadira2006). Gulick went further and proposed, as a solution to this Phosphate Problem, that lower oxidation state phosphorus oxyacids, specifically H-phosphonic (H3PO3) and H-phosphinic acids (H3PO2; Fig. 1), would have been more thermodynamically stable within a reducing Hadean environment (Gaidos et al. Reference Gaidos, Nealson and Kirschvink1999), more soluble in an early-Earth ocean and, given their position in the thermodynamic Frost diagram (Fig. 1), serve as more effective phosphorylating agents for prebiotic organics than orthophosphate. The principal difficulties with Gulick's proposal lay in: (i) identifying effective sources of phosphonates and phosphinates for the early Earth and (ii) delineating systematically what role, if any, such species may have played in either prebiotic chemistry or in development of a burgeoning biosphere. Subsequently, Cooper et al. (Reference Cooper, Onwo and Cronin1992, Reference Cooper, Thiemens, Jackson and Chang1997) reported alkylphosphonic acids in the Murchison meteorite and Schwartz and co-workers (Schwartz Reference Schwartz1995, Reference De Graaf, Visscher and Schwartz1997; De Graaf et al. Reference De Graaf, Visscher and Schwartz1995, Reference De Graaf, Visscher and Schwartz1997, Reference De Graaf, Visscher and Schwartz1998; De Graaf & Schwartz Reference De Graaf and Schwartz2000) have published widely on the potential of H-phosphonic acid to prebiotic chemistry. Recent results from both our own laboratories (Pasek & Lauretta Reference Pasek and Lauretta2005; Bryant & Kee Reference Bryant and Kee2006) have identified a readily available exogenous source of low-oxidation state phosphorus to the early Earth in the form of iron meteorites. These studies revealed the ease with which schreibersitic, (Fe,Ni)3P, inclusions within iron meteorites afford principally H-phosphonic acid under thermal (Pasek & Lauretta Reference Pasek and Lauretta2005) and H-phosphinic acid under photochemical conditions (Bryant & Kee Reference Bryant and Kee2006).

Fig. 1. Frost diagram of some geologically and biologically relevant phosphorus species under acidic conditions (pH=0; T=298 K; 1 atm pressure and unit solution activity).
Since Precambrian oceans were both richer in lower-oxidation state species (Brocks et al. Reference Brocks, Love, Summons, Knoll, Logan and Bowden2005) including metal salts such as Fe2+ and, at ca. 70°C, significantly warmer than present day oceans (Robert & Chaussidon Reference Robert and Chaussidon2006), a considerable fraction of the meteoritic and cometary in-fall during the late heavy bombardment (Chyba & Sagan Reference Chyba and Sagan1992) would have been exposed to a hot, reducing, aqueous saline environment. Iron meteorites would have been a major component of this in-fall and, given the consistent presence of schreibersite inclusions within such bodies, it is likely that a proportion of that estimated 109 kg yr−1 of meteoritic phosphorus (Pasek & Lauretta Reference Pasek and Lauretta2008) would have experienced anoxic, sub-oceanic aqueous modification in the absence of solar radiation.
Here we report facile phosphorus redox chemistry of schreibersite-containing iron meteorites and their possible impact upon the evolution of early Earth environments. As part of this, we describe the first application of scanning Kelvin probe techniques to the study of meteoritic chemical corrosion.
Experimental methods
Leaching of phosphorus from the Seymchan pallasite
The Seymchan meteorite is an anomalous main-group pallasite meteorite (van Niekerk et al. Reference Van Niekerk, Greenwood, Franchi, Scott and Keil2007) from the Magadan region of Russia. Seymchan is a phosphide-rich meteorite; total phosphorus abundances suggest 0.22 wt% total phosphorus (Kirova & Dyakonova Reference Kirova and Dyakonova1972). However, this estimate likely excludes macroscale phosphide inclusions, which are common in the meteorite. Seymchan metal shavings were purchased as waste material left over from meteorite cuttings, and 7.785 g were washed with acetone and isopropanol (70:30 v/v isopropyl alcohol to water), then these shavings were placed in 25 cm3 of 0.025 M Na4EDTA solution and stirred for two weeks in a round bottomed flask sealed with parafilm. An aliquot of this solution (10 cm3) was removed and brought to a pH of 13 by the addition of 10 M NaOH. A red precipitate settled to the bottom of the flask; the solution retained a blue colour (likely dissolved Ni2+). The solution was analysed using 31P NMR on a Varian 300 four-nucleus probe Fourier transform nuclear magnetic resonance (FT-NMR) spectrometer at 121.43 MHz and 24.5°C for 3424 scans following prior work (Pasek & Lauretta Reference Pasek and Lauretta2005; Pasek et al. Reference Pasek, Dworkin and Lauretta2007).
Fabrication of mineral electrodes
Fe3P powder (1 g, Alfa Aesar, 95.5%, −40 mesh powder) was compressed under more than 10 tonnes of pressure in a 1 cm diameter circular steel die followed by sintering on a bed of graphite at 1000°C for 1 hr within an alumina crucible. An electrical connection was made using conducting silver-loaded epoxy cement to a copper wire, sealed using either beeswax or an epoxy resin. The surfaces were polished using light emery paper prior to beginning electrochemical studies.
Measurements of static electrode potentials
Static potentials were measured at 298 K against the saturated calomel electrode (SCE), stabilized and measured after 5 minutes at high impedance with a Black Spur DT-100 multimeter on the 2000 mV setting. De-gassed, N2-saturated, aqueous brine solution (3.5% w/w NaCl) was used for each and pH measurements were performed with a pre-calibrated Schochem pH meter. Some care must be exercised here in extrapolating these models to actual meteorite samples in that static reduction potentials are likely to be altered by inclusion of the more noble metal nickel. The Fe3P electrode was biased as an anode against a copper (E or=+0.36 V) cathode, the latter being used in place of an Fe:Ni matrix. Being a pure element rather than an alloy, the copper displayed a more stable potential than an actual meteoritic sample such as Nantan (ca. +0.1–0.3 V), whose potential would be affected by electrochemical interactions occurring between the different elements on its surface. The salt bridge was fabricated by addition of agar to warm (ca. 80°C) saturated aqueous KCl and the resulting mixture introduced under reduced pressure into an opaque plastic tube (20 cm length; 0.5 cm outer diameter) and allowed to cool to room temperature whereupon gel formation occurred within the tube.
Galvanic cell
A Galvanic cell comprising a pressed disc of Fe3P as the anode (vide supra) and copper wire as the cathode was constructed with both cell compartments containing 50 cm3 of degassed 3.5% w/w aqueous NaCl connected by a saturated KCl-agar salt-bridge. The cell was run for 48 h at 300 K, under a dynamic atmosphere of dinitrogen, against a peak potential of 350 mV monitored on a Black Spur DT-100 multimeter. Subsequently, the salt solution from the anodic chamber was collected, treated with in excess of 1 M aqueous Na2S solution and the resulting FeS precipitate centrifuged to compactness before being filtered. The resulting filtrate was evaporated under reduced pressure and the resulting solid dissolved in D2O prior to analysis via 31P{1H}-NMR spectroscopy on a Bruker ARX250 FT-NMR system operating at 300 K and 101.26 MHz for phosphorus.
Scanning Kelvin probe
The scanning Kelvin probe (SKP) experiments were carried out with a Uniscan SKP 100 instrument fitted with a platinum tip. The gap between the polished specimen and the probe tip was approximately 100 μm. Area scans were conducted using a range of data points per line between 8 and 512 to determine an optimum resolution; no improvement in the output obtained was observed above 128 points per line over the area scanned in this study, so this setting was used to gather data. The instrument was calibrated against a number of known redox potentials, in line with the published method (Stratmann & Streckel Reference Stratmann and Streckel1990), so that the SKP outputs could be expressed in terms of corrosion potentials. The scans were carried out in Step-Scan mode with the instrument enclosed in a chamber to reduce the influence of temperature and humidity fluctuations over the duration of the experiment.
Computational methods
All calculations were carried out using the Gaussian 03 suite of programs (Frisch et al. Reference Frisch2004). Geometry optimizations and vibrational frequency calculations were performed at the B3LYP/cc-pVTZ level.Footnote 1 A scale factor of 0.9854 was used for zero-point energy corrections. The accurate energetics in the gas phase were evaluated using G3X model chemistry, which approximates the electron correlation level of QCISD(T,Full)Footnote 2/G3XLarge (Curtiss et al. Reference Curtiss, Redfern, Raghavachari and Pople2001). A CPCM model was used to account for the solvation effect (Barone & Cossi Reference Barone and Cossi1998; Cossi Reference Cossi2003).
Results and discussion
A typical schreibersite inclusion, located within the type IIIB Bear Creek iron meteorite, is illustrated in Fig. 2. The back-scattered electron scanning electron microscopy (SEM) image (Fig. 2(a)) shows the inclusion outlined clearly against an Fe:Ni matrix, along with distinctive corrosion pits within the matrix (black regions on the right-hand side of Fig. 2(a)).

Fig. 2. (a) SEM secondary electron image (20 kV) of schreibersite inclusion in the type IIIB Bear Creek meteorite. (b) EDX map at phosphorus K α(2.0137 KeV). (c) EDX map at oxygen K α(0.5249 KeV).
Energy dispersive X-ray (EDX) maps at oxygen K α(0.5249 KeV; Fig. 2(b)) and phosphorus K α(2.0137 KeV; Fig. 2(c)) frequencies respectively clearly show that the inclusion is phosphorus-rich and also that the mineral boundary between the inclusion and matrix appears especially susceptible to oxidation (vide infra) with clear oxide formation at this point. The significance of these surface analyses to potential redox chemistry within an early Earth environment is emphasized by Vickers microhardness data on a range of schreibersite inclusions in various iron meteorites. Our measurements reveal that Vickers numbers (HV) for schreibersite inclusions are significantly larger than those measured for matrix material (taenite and/or kamacite), which is consistent with the former being significantly more brittle as found in other phosphide minerals (Pratesi et al. Reference Pratesi, Bindi and Moggi-Cecchi2006) and hence more susceptible to fissure than the matrix (Table 1).
Table 1. Vickers microhardness numbers (HV) for both schreibersite inclusions (located and analysed by SEM/EDX) and the Fe:Ni matrix in several iron meteorites. 1=Bear Creek, 2=Toluca and 3=TomBigBee River. Meteorites samples courtesy of the Natural History Museum
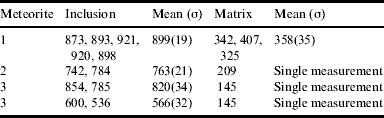
Thus, one may envisage an iron meteorite that fissures upon impact to expose the hard, brittle schreibersite inclusions to a warm, moist environment. Furthermore, given the inevitable compositional differences between the electrically conducting Fe:Ni matrix and the metal phosphide inclusion, one might anticipate associated differences in electrode potential across inclusion boundaries leading to anoxic electrochemistry in the presence of a suitable electrolyte solution. Recognizing this, we have sought to model the electrochemical behaviour of schreibersite inclusions by fabricating electrodes from commercial Fe3P powder and measuring static electrochemical potentials against the saturated calomel electrode, comparing these subsequently to measurements of pure iron and an Fe:Ni matrix from the Nantan meteorite (Table 2).
Table 2. Static electrode potentials (in volts) of iron-based electrodes versus saturated calomel electrode (SCE); de-gassed, N2-saturated, aqueous brine (3.5% w/w NaCl) at 298 K. Top value versus SCE; lower value versus SHE (standard hydrogen electrode). Static potentials did not reveal significant differences over the pH range 5.1–8.3. aMg2+ and Fe2+ as sulphates, Ca2+ as nitrates all at 1 M metal concentration. bMeasurements were performed in duplicate on two separately manufactured Fe3P electrodes and with both iron and Nantan electrodes: standard deviations in parentheses. cIn the form of pure iron wire, folded and pressed into a thin sheet ca. 2×10 mm2. dStandard reduction potential for the Fe2+(aq)+2e=Fe(s) half cell versus SHE is −0.44 V. eValues fluctuated too widely for confidence. fSample provided by British Jurassic Fossils

The more negative the potential of an electrode versus SCE, the more anodic that electrode and the greater the potential for anodic oxidation of the electrode material. Thus, iron in Fe3P appears to be more susceptible to anodic oxidation to Fe2+ than pure iron in the presence of solutions containing significant (1 M) concentrations of Mg2+ and Ca2+, a result that appears to have some precedent (Tanabe et al. Reference Tanabe, Shibuya, Kobayashi and Misawa1997). In aqueous saline solution containing 1 M Fe2+ however, it appears less clear whether Fe3P or pure Fe is the more sensitive towards anodic oxidation. In each case, the Fe3P electrode appeared more susceptible to anodic oxidation than an electrode based on the Nantan meteorite.
A more direct technique for exploring corrosion behaviour of electrochemically differentiated alloys exploits the SKP, a technique well-established in metallurgy (Cheran et al. Reference Cheran, Johnstone, Sadeghi and Thompson2007; Bozec et al. Reference Bozec, Persson, Nazarov and Thierry2002), but previously unreported for studying iron meteorite samples. In Fig. 3(a) an optical image of a section of the Seymchan pallasite clearly shows the bright inclusion of schreibersite and beneath it (Fig. 3(b)) SEM images and EDX analyses of both the inclusion (left) and the matrix (right) clearly confirm the presence of phosphorus in the form of phosphide rather than orthophosphate in the inclusion. The hydrothermal treatment of Seymchan shavings has been confirmed by 31P-NMR spectroscopy to afford leached phosphorus as hypophosphate (4% of solution P), orthophosphate (34%), phosphite (49%) and pyrophosphate (13%; Fig. 4), commensurate with those found under anoxic conditions for the Nantan meteorite (Bryant & Kee Reference Bryant and Kee2006).

Fig. 3. (a) Optical image of a ‘twin-fingered’ schreibersite inclusion (ringed) within a sectioned sample of the Seymchan pallasite. (b) SEM images and EDX data of one point within one of fingers of the ‘twin-fingered’ region (left) and matrix (right). (c) 2D-SKP map of highlighted rectangular region (12 mm×9 mm) clearly showing the twin-fingered motif as a lighter colouration against a blue background (colour scale indicated beneath figure).

Fig. 4. Seymchan phosphorus speciation NMR spectrum. Peaks correspond to hypophosphate (P2O64−, δ ca. 14 ppm), orthophosphate (PO43−, δ ca. 6 ppm), phosphite (HPO32−, δ ca. 4 ppm), and pyrophosphate (P2O74−, δ ca. −4 ppm).
A SKP map of the region highlighted in Fig. 3(a) is reproduced in Fig. 3(c). The SKP technique measures differences in surface work functions that reveal themselves as colour differences in the two-dimensional map. Once calibrated, the SKP can be used to measure electrochemical potentials across a surface and subsequently corrosion currents. Analysis of the SKP outputs for the inclusion mapped in Fig. 3(c) indicates a potential difference between the matrix mineral and the schreibersite inclusion of ca. 400 mV with the inclusion being electrochemically more noble than the matrix; hence the matrix would tend to corrode preferentially as the anodic part of a galvanic cell. Further studies are on-going to better quantify corrosion currents expected within different environments and these will be published in due course. This 400 mV potential difference is consistent with higher proportions of the more electrochemically noble nickel within the inclusion compared to the matrix. Since anodic oxidation events must be balanced by cathodic reduction, these results suggest that preferential anodic oxidation of the matrix may drive cathodic reduction at the matrix–schreibersite interface. Indeed, the observation of enhanced oxide deposits at the matrix–inclusion interface of a schreibersite inclusion in Bear Creek (Fig. 2(c)) would tend to support such a hypothesis.
This would be consistent with oxygen reduction via O2(g)+2H2O(l)+4e=4OH−(aq), which is the predominant cathodic corrosion process in natural aerobic conditions. Whilst anodic oxidation of iron in Fe3P and meteoritic schreibersite to afford Fe2+ is unsurprising, the more interesting chemical phenomenon surrounds associated redox chemistry of the phosphorus. Thus, mindful of the fact that eventually all iron within iron meteorites will tend thermodynamically towards bulk anodic oxidation, we have examined electrochemical cells with an Fe3P electrode as anode biased against a copper cathode via a KCl/agar salt bridge (Fig. 5(a)).

Fig. 5. (a) Anaerobic Galvanic cell comprising pressed disc Fe3P. (b) As anode and copper wire cathode in separate chambers (50 cm3 3.5% w/w aq. NaCl) connected by a KCl-agar salt bridge. (c) 31P{1H}-NMR (H2O; 300 K) of the anodic compartment of the cell in Fig. 4(a) identifying the key P-containing oxyacid products; H3PO2 (91%); H3PO3 (7%); H3PO4 (2%).
A steady current flow of ca. 3 μA against a peak potential difference of 350 mV was noted and maintained within the cell for 48 h at 300 K under an atmosphere of dinitrogen in 3.5% brine. After this time interval 31P{1H}-NMR spectroscopy identified the dominant component in solution to be H-phosphinic acid (H3PO2; Fig. 4(c)) by comparison to an authentic sample. This, we believe, is a highly significant result as it demonstrates that anoxic corrosion of phosphide inclusions within electrochemically differentiated iron meteorites readily affords highly-reactive, water-soluble oxyacids of phosphorus via anodic oxidation (Pasek & Lauretta Reference Pasek and Lauretta2005; Bryant & Kee Reference Bryant and Kee2006).
A corrosion current of 3 μA coupled with an anodic surface area πr 2 (where the electrode radius r=5×10−3 m) of 7.85×10−5 m2 should afford a corrosion current density i corr of 3×10−6/7.85×10−5=0.038 A m−2 or 0.0038 mA cm−2. As a current density of 1 mA cm−2 equates to an iron corrosion rate of ca. 249 g m−1 day−1, a current density of 0.0038 mA cm−2 should provide a mass loss of ca. 0.95 g m−2 day−1. After an experiment time of 7 days (anoxic, currents of ca. 2–3 μA) we observe total dissolved Fe at ca. 1.7 ppm and P at ca. 3 ppm via ICP-MS (inductively coupled plasma–mass spectrometry) which, if we assume that what we are observing is a lower limit to P concentration, would equate to 3 mg l−1. In a total saline solution of 100 cm3, this would afford 0.3 mg of P in solution from a 1 g anodic disc of Fe3P containing ca. 17% P w/w, which is a P-corrosion rate of ca. 0.2% per week or 10% per year. Even though the conditions explored above are rather staged from a prebiotic context, exposed meteoritic schreibersite inclusions may be expected to release significant fractions of their P-content via anoxic Galvanic corrosion in salt water on relatively short geological timescales. Indeed, both previous experiments (Bryant & Kee Reference Bryant and Kee2006) and those described here show that meteoritic phosphides release reduced P on geologically short timescales. In general, phosphides such as schreibersite will be exposed on about 5% of the surface area of an iron meteorite. Other extraterrestrial materials, such as interplanetary dust particles, have huge surface area to volume ratios, so it may be reasonable to assume that these particles behave similarly to our experimental system. Given the quantities of siderophilic P calculated to have impacted the Earth during the late heavy bombardment (Macia Reference Macia2005), coupled with the high fraction of H-phosphinic acid in the corrosion P-products, we anticipate that highly water-soluble and chemically-reactive phosphorus salts were readily and widely available to prebiotic chemistry upon the early Earth. If we assume the duration of the heavy bombardment period to be 107–108 years, we estimate that approximately 1015–1017 kg of reduced phosphorus were delivered to the early Earth (Macia Reference Macia2005). This comprises 0.1–10% of the total crustal phosphorus. If all of this reduced phosphorus were dispersed in the oceans of the early Earth, the total concentration of reduced P in the early oceans would be between 10 μM and 1 mM, a factor of 10 or 1000 times the present concentration, respectively.
Thermodynamic considerations and concluding remarks
Terrestrial phosphorus geo- and biochemistry have traditionally focused almost exclusively upon fully oxidized orthophosphate and condensed polyphosphate materials. The role of phosphorus in both these spheres is now being rewritten (Pasek Reference Pasek2008) to include low oxidation state species with concomitant phosphorus redox chemistry, which is a novel concept in the chemical evolution of phosphate-based life. In-fall of extraterrestrial material such as meteorites and interstellar dust particles during the late heavy bombardment brought significant quantities of reduced oxidation-state phosphorus to the early Earth in the form of siderophilic minerals such as schreibersite. Our work here shows that electrochemical transformations of phosphide minerals under anoxic conditions afford water-soluble and chemically-reactive phosphorus compounds including H-phosphinic acid (H3PO2). In addition, we report the first application of the scanning Kelvin probe technique to map the electrochemical potentials of an iron meteorite surface, clearly revealing differences between schreibersite inclusions and the surrounding Fe:Ni matrix. We will be extending this technique to a range of other meteorite types with a view to probing corrosion behaviours in different planetary environments.
We have reported briefly on the ease with which H-phosphinic acid undergoes transformations to organophosphorus compounds in the presence of aqueous alcohols and photochemical irradiation (Bryant & Kee Reference Bryant and Kee2006) and, whilst more detailed investigation of such chemistry will be described elsewhere, we feel it is apposite to some of the thermodynamic implications of phosphorus redox chemistry on the early Earth.
In Table 3 selected thermodynamic parameters for a range of processes of relevance to phosphorus chemistry under putative early Earth conditions are summarized. Whilst all processes modelled are thermodynamically favourable in the gas phase as judged by ΔG o(298) values, condensed phase solution parameters are more revealing. Processes 1 and 2 represent alcoholysis reactions of orthophosphoric (1) and H-phosphonic (2) acids respectively, both being endothermic by ca. 11–12 kJ mol−1, consistent with the expected relatively poor abilities of both P-species to phosphorylate simple organic alcohols. Nature has circumvented this problem by exploiting catalysed hydrolysis of the (P–O–P) bond within condensed polyphosphates such as adenosine triphosphate (ATP) (Westheimer Reference Westheimer1987). Hydrolysis of the parent derivative, pyrophosphate, process (3) is thermodynamically favourable by ca. 55 kJ mol−1 whilst alcoholysis reactions (exemplified by process (4)) are similarly exothermic. A model for the chemical evolution of condensed polyphosphates is high on the list of key problems and one that is currently being addressed (Pasek et al. Reference Pasek, Kee, Bryant, Pavlov and Lunine2008), but we believe it is possible that lower oxidation state phosphorus oxyacids could have provided a significantly more favourable thermodynamic field within which to pursue prebiotic chemistry. Processes (5)–(9) represent oxidation of H-phosphinic ((5) and (8)) and H-phosphonic systems ((6), (7) and (9)) with concomitant reduction of H+ to H2; these are processes that have the potential to release significant free energy to drive other, potentially endothermic chemical reactions. Preliminary calculations suggest, however, that the activation barrier to process (5), hydrolytic oxidation of H3PO2 to H3PO3 and H2, is likely to be insignificant in solution given a gas-phase value of ca. 250 kJ mol−1, but we envisage that the intervention of mineral and/or metal surfaces would reduce such barriers significantly. The coupled oxidation of H3PO2 and H3PO3 with dioxygen as oxidant provides, not unsurprisingly, considerably more energy (processes (10) and (11)), which may have had a role to play in the earliest stages of chemical evolution given the identification of anoxic bacteria capable of using phosphonate oxidation to drive sulphate reduction (Schink & Friedrich Reference Schink and Friedrich2000). Finally, we identify (P–H) insertion processes of H-phosphinic acid (process (12)) as being especially interesting as they combine significant energy production of ca. 75 kJ mol−1 with organic-functionalized phosphorus products of a type that are both chemically robust and have the potential for further chemical transformation. Indeed, insertion into the second (P–H) bond of a mono-functionalized organo-H-phosphinate affords a bi-functionalized phosphinate diester with release of modest free energy. Bi-functional derivatives of this type may open a potential prebiotic route to phosphinate polymers, which is a line of investigation we are currently actively pursuing in our laboratories.
Table 3. Calculated thermodynamic properties at G3 level for gas phase, with CPCM model correction for solution phase. All are based on B3LYP/cc-pVTZ geometries and ZPE (zero point energy) corrections

Acknowledgements
We thank the Leverhulme Trust (grant F00/122/X), the EPSRC (grant EP/F042558/1) and NASA (Exobiology and Evolutionary Biology grant NNX07AU08G) for financial support, the Natural History Museum, London for the generous loan of sectioned meteorite samples (loan 249), Jon Wright (Sheffield Hallam University) for microhardness measurements and Martin Huscoft (University of Leeds) for ICP-MS analyses.








