1. Introduction
During Phanerozoic times, the concentrations of the main ions present in ocean water as Cl−, SO4 2−, HCO3 −, Na+, K+, Mg2+ and Ca2+ have varied significantly. The variation of the ion concentration has been investigated over decades and has been the subject of continuous debates (Hardie, Reference Hardie1996; Kovalevich, Peryt & Petrichenko, Reference Kovalevich, Peryt and Petrichenko1998; Lowenstein et al. Reference Lowenstein, Hardie, Timofeeff and Demicco2003; Holland, Reference Holland2005; Lowenstein et al., Reference Lowenstein, Kendal, Anbar, Holland and Turekian2014; Algeo et al. Reference Algeo, Luo, Song, Lyons and Canfield2015). For example, Permian seawater had a SO4 2− concentration similar to that of modern seawater. Sulfate-rich marine waters were generally an exception in the history of the Earth and are restricted to late Precambrian (Vendian), Pennsylvanian–Triassic and Miocene to Quaternary times (Hardie, Reference Hardie1996). During such periods, potash deposits formed, which are characterized by the presence of MgSO4 salts, such as polyhalite and kieserite. These periods have alternated with periods characterized by the formation of KCl salts, such as sylvite (KCl), as found in the Cambrian through Mississippian, and Jurassic through Palaeogene.
Sulfate minerals contain besides elements such as calcium, strontium, barium, magnesium and potassium also sulfur, oxygen and, in variable amounts, water. Sulfate evaporitic deposits represent important archives of Earth history, their formation indicating particular basinal and climatic conditions. Sulfur and oxygen stable isotope compositions of evaporites are controlled by global cycles through geological times as well as by local environmental factors. Thus, isotopic investigations on sulfur and oxygen from sulfate accumulations combined with mineralogical investigations may offer information regarding the origin of fluids, inflows or restricted conditions, evaporative effects, recrystallization and bacterial processes (Longinelli & Craig, Reference Longinelli and Craig1967; Holser et al. Reference Holser, Kaplan, Sakai and Zak1979; Ohmoto, Reference Ohmoto, Valley, Taylor and O'Neil1986; Hałas, Reference Hałas1987; Raab & Spiro, Reference Raab and Spiro1991; Machel, Krouse & Sassen, Reference Machel, Krouse and Sassen1995; Seal, Alpers & Rye, Reference Seal, Alpers, Rye, Alpers, Jambor and Nordstrom2000; Canfield, Reference Canfield, Valley and Cole2001; Kucha et al. Reference Kucha, Schroll, Raith and Hałas2010; Peryt, Hałas & Hryniv, Reference Peryt, Hałas and Hryniv2010; Boschetti et al. Reference Boschetti, Cortecci, Toscani and Iacumin2011; García-Veigas et al. Reference García-Veigas, Cendón, Pueyo and Peryt2011).
The sulfur and oxygen isotopic composition of sulfate accumulations has varied in time and space. For example sulfur-isotope age curves for marine sulfate include the work of Nielsen (Reference Nielsen1965), Holser (Reference Holser1977), Claypool et al. (Reference Claypool, Holser, Kaplan, Sakai and Zak1980) and Strauss (Reference Strauss1997). Sulfur-isotope age curves for structurally substituted sulfate in marine carbonate were also determined (Kampschulte & Strauss, Reference Kampschulte and Strauss2004; Prokoph, Shields & Veizer, Reference Prokoph, Shields and Veizer2008), as well as a higher resolution record for Cretaceous barite accumulations (Paytan et al. Reference Paytan, Kastner, Campell and Thiemens2004).
As the present study refers to evaporites of Late Permian – Triassic age, the general trends for this period of time will be summarized. The δ34S values of sulfates decreased towards the Late Permian (Nielsen, Reference Nielsen1965; Holser, Reference Holser1977; Claypool et al. Reference Claypool, Holser, Kaplan, Sakai and Zak1980); a similar trend was also encountered for the δ18O values of sulfates (Claypool et al. Reference Claypool, Holser, Kaplan, Sakai and Zak1980). The isotopic composition of sulfur from oceanic sulfate is mainly controlled by a balance of the erosion input, mainly shales, as well as bacterial reduction and formation of sulfides. The low δ34S values of Upper Permian sulfates are associated with the predominance of sulfur in oxidized phases, as gypsum or polyhalite. During the same time, carbon burial and the oxygen content of the atmosphere were high. Low isotopic values characteristic for marine sulfate of Late Permian age increased sharply during the formation of the Triassic Röt evaporites (Nielsen, Reference Nielsen1965; Holser, Reference Holser1977; Claypool et al. Reference Claypool, Holser, Kaplan, Sakai and Zak1980). Besides general trends, significant isotopic variations of evaporitic sulfate as high as 3‰ were observed in different places within a given lithostratigraphic unit both vertically and horizontally, depending on a variety of depositional factors such as different degrees of restriction of the evaporitic basin to the open ocean, bacterial sulfate reduction in local evaporating basins and variation in terrigenic input (Claypool et al. Reference Claypool, Holser, Kaplan, Sakai and Zak1980; Cortecci et al. Reference Cortecci, Reyes, Berti and Casati1981; Fanlo & Ayora, Reference Fanlo and Ayora1998; Longinelli & Flora, Reference Longinelli and Flora2007). Post-depositional isotopic variations may be caused by the exchange of sulfate with water under higher temperature conditions that should affect the oxygen isotopes only. If thermal overprinting occurs in a practically closed system with a low water–rock ratio, the isotopic effects should be small for sulfur. Therefore, sulfur isotope composition is largely preserved during thermal overprinting, unless external fluids are involved.
Sulfur isotope compositions were earlier analysed for sulfates from the Permian–Triassic deposits of the Northern Calcareous Alps (NCA) (Pak, Reference Pak1974, Reference Pak1981; Spötl & Pak, Reference Spötl and Pak1996). As sulfate oxygen data have been missing up to now, the aims of the present study are to produce a new dataset of integrated sulfur and oxygen data for Permian–Triassic deposits situated in different units of the Eastern Alps. The data will provide information regarding the conditions of formation, regional extent and the subsequent history of the sulfate deposits.
2. Geology of investigated sulfate deposits
The Eastern Alps are characterized by the presence of three main tectonic units, the Lower, Middle and Upper Austroalpine, which overlie the Penninicum (Tollmann, Reference Tollmann1977). The Upper Austroalpine unit consists of the NCA overlying the Greywacke zone and corresponding to the Graz Palaeozoic, Murau Palaeozoic and Gurktal nappe, with evaporitic rocks lacking in the later ones. The Mesozoic NCA belt is detached and thrust along the evaporitic Upper Permian to Lower Triassic Haselgebirge Formation. The sedimentation started in Late Carboniferous or Early Permian times, the youngest sediments being of Eocene age. The NCA are divided into the Bajuvaric, Tirolic and Juvavic nappe complexes. The evaporitic Haselgebirge Formation occurs in connection with the Juvavic nappe complex. Some evaporites are also situated in the Tirolic units (Leitner & Neubauer, Reference Leitner and Neubauer2011). The Haselgebirge Formation consists mainly of rock-salt, shales, gypsum and anhydrite and includes the oldest sediments of the NCA. The age of the Haselgebirge Formation, established by using spores and geochronological data, is Permian to Early Triassic (Klaus, Reference Klaus1965; Tollmann, Reference Tollmann1977; Pak, Reference Pak1974, Reference Pak1981; Pak & Schauberger, Reference Pak and Schauberger1981; Flügel & Neubauer, Reference Flügel and Neubauer1984; Schauberger, Reference Schauberger1986; Spötl, Reference Spötl1988 a; Spötl, Reference Spötl1989 a; Weber, Reference Weber1997; Schorn et al. Reference Schorn, Neubauer, Genser and Bernroider2013; Leitner et al. Reference Leitner, Neubauer, Genser, Borojevic-Sostaric, Rantitsch, Jordan, Mark and Verati2013). Palaeomagnetic investigations of Triassic to Lower Cretaceous deposits from the NCA indicate remagnetization during nappe stacking and folding events related to Alpine orogenesis (Pueyo et al. Reference Pueyo, Mauritsch, Gawlick, Scholger and Frisch2007).
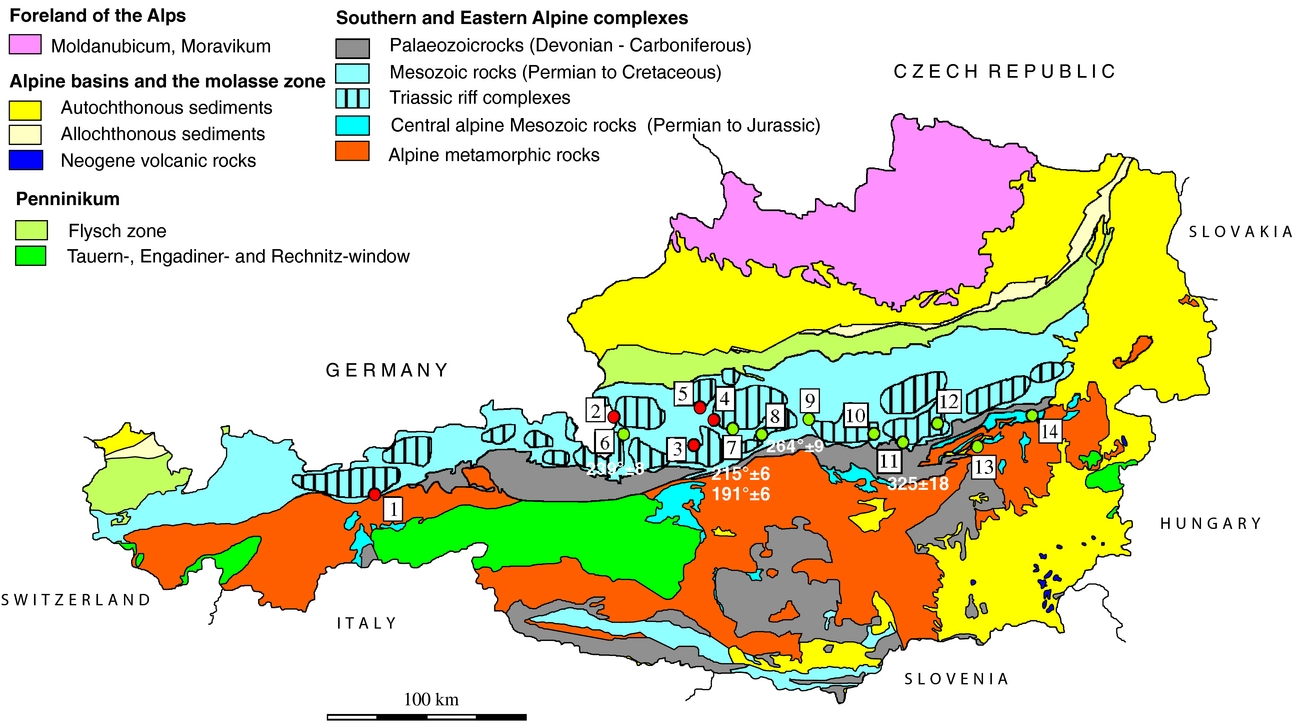
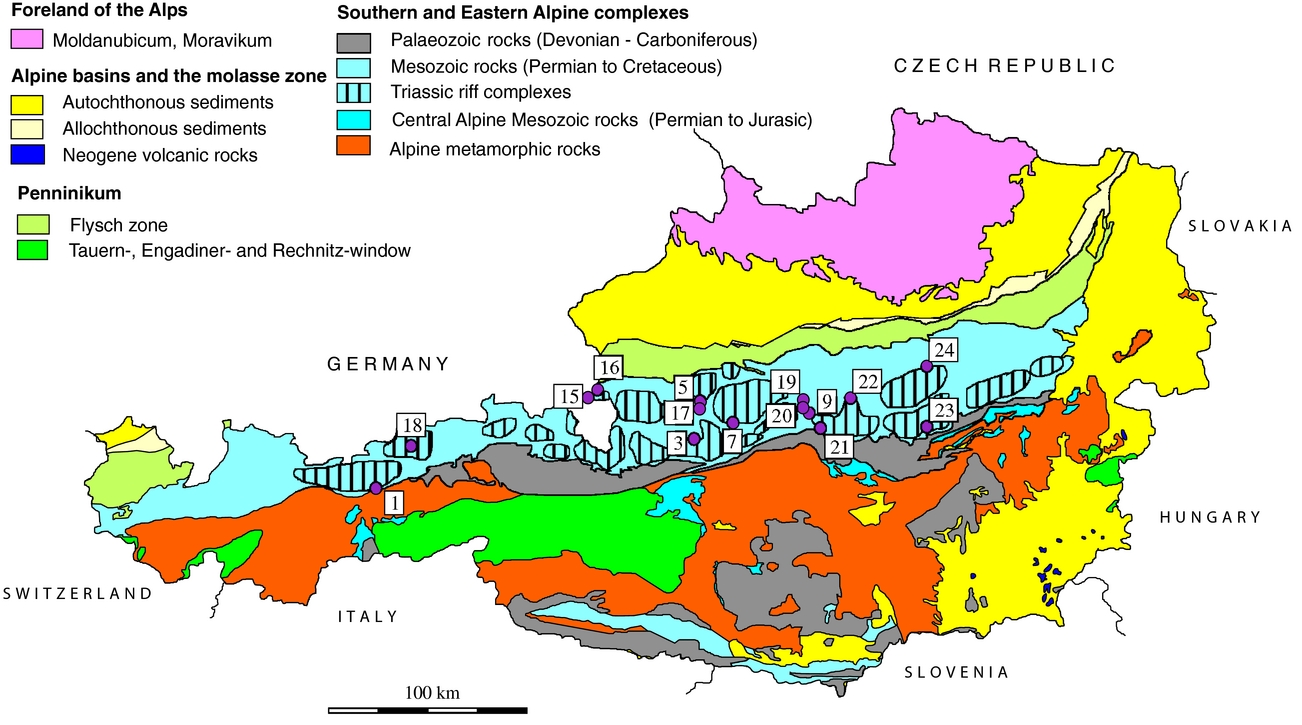
In this study we investigated sulfates from halite and gypsum deposits; the regional distribution of samples is given in Figure 1. The deposits from where samples were taken are briefly presented below; the number corresponding to Figure 1 is indicated in brackets.
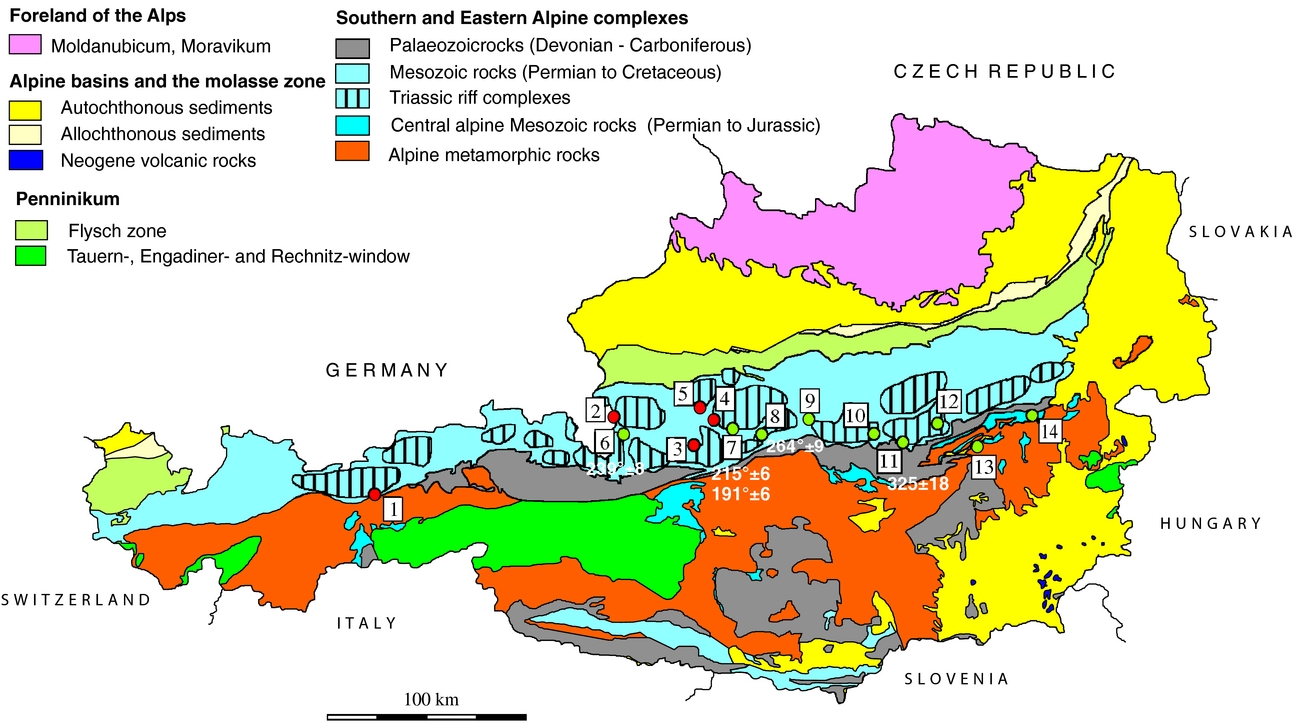
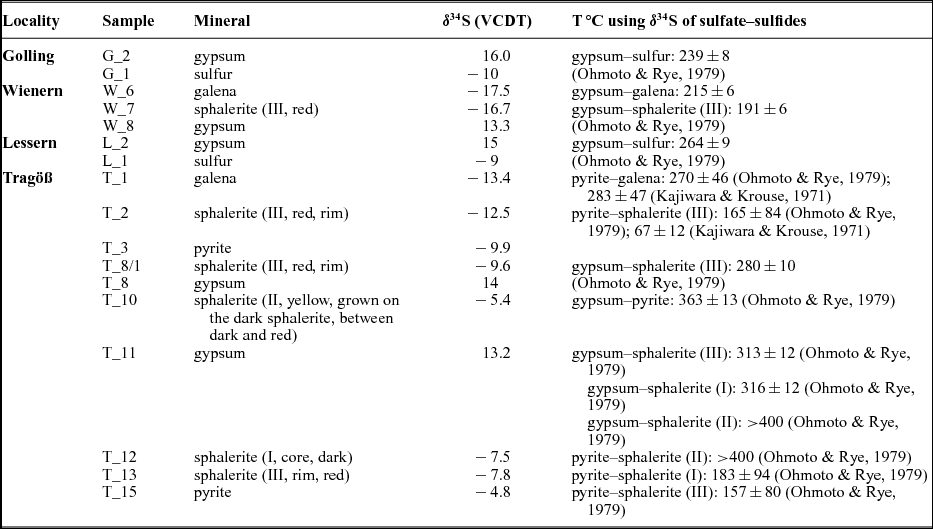
Figure 1. Geological overview of the Eastern Alps showing the distribution of investigated sulfate accumulations. Distribution of calculated temperatures using sulfur isotope composition (Table 4). Salt: 1 – Hall in Tirol; 2 – Hallein; 3 – Hallstatt; 4 – Altaussee; 5 – Bad Ischl. Gypsum deposits, Northern Calcareous Alps: 6 – Golling; 7 – Wienern; 8 – Lessern; 9 – Unterlaussa; 10 – Wildalpen; 11 – Tragöß; 12 – Seewiesen. Gypsum deposits, Central Alpine Mesozoic: 13 – Stanz; 14 – Göstritz.
2.a. Salt deposits of the Northern Calcareous Alps sampled for this study
Hall in Tirol (1) is the westernmost salt-mine in the Eastern Alps and was closed in the 1960s after ~700 years of mining. It has an E–W extension of ~1 km. The deposit is situated at the base of the Tirolic unit next to the underlying Bajuvaric unit. The hosting rock, the so-called Haselgebirge, is a tectonic breccia of shales, sulfate rocks, rock-salt and sandstones. Sulfur isotope and palynological studies show a Late Permian age (Klaus, Reference Klaus1965). A second evaporitic event occurred during Anisian time and is represented by dolomite/anhydrite rocks (Pak, Reference Pak1974; Spötl, Reference Spötl1988 a,b; Spötl, Reference Spötl1989 b; Weber, Reference Weber1997; Leitner & Neubauer, Reference Leitner and Neubauer2011).
The Hallein/Dürnberg (2) deposit is interconnected with the salt deposit in Berchtesgaden, Germany. The ore body extends from SW–NE with a length of ~ 7 km and is up to 2 km wide. The base of the salt body is ~ 1 km deep below the surface. The deposit is situated between a lower Tirolic and an upper Juvavic nappe and has a Late Permian age (Gawlick et al. Reference Gawlick, Lein, Piros and Pytel1999). The salt incorporated Lower Triassic to Jurassic host rocks (Leitner & Neubauer, Reference Leitner and Neubauer2011).
The Hallstatt (3) and Dürrnberg deposits have been mined since the Bronze Age. The Hallstatt deposit is also well known as it is named after the Hallstatt culture stage. The deposit extends E–W and is 3 km in length and 600 m in width. Host-rock incorporations include besides Permian volcanic rocks also Triassic to Jurassic rocks (Gawlick et al. Reference Gawlick, Lein, Schlagintweit, Suzuki and Wegerer2001; Leitner & Neubauer, Reference Leitner and Neubauer2011).
Altaussee (4) is an actively mined rock-salt deposit which covers an area of ~ 2.3 km2. A c. 1 km deep drilling did not reach the bottom of the deposit. The only host rocks incorporated in the rock-salt are of the Triassic Hallstatt formation (Leitner & Neubauer, Reference Leitner and Neubauer2011). The deposit is covered by the Middle Jurassic Sandling-Alm Formation (Gawlick, Schlagintweit & Suzuki, Reference Gawlick, Schlagintweit and Suzuki2007).
Bad Ischl is a rather small (~ 0.5 km2) rock-salt deposit. The deposit is jammed between Triassic, Jurassic and Cretaceous rocks. Rocks incorporated to the rock-salt are Permian volcanic rocks, Triassic rocks from the Hallstatt formation and Cretaceous sandstones and conglomerates (Leitner & Neubauer, Reference Leitner and Neubauer2011).
2.b. Gypsum deposits of the Northern Calcareous Alps sampled for this study
Numerous, partly mined sulfate deposits occur along with the rock-salt deposits of the NCA. Besides the major phases of gypsum and anhydrite, a large number of sulfide phases like galenite, sphalerite, pyrite, lead-arsen-sulfosalts, tennantite and native sulfur a.s.o. are reported frequently. Carbonates such as magnesite, dolomite and calcite can be found either as singular crystals (Kirchner, Reference Kirchner1987; Niedermayr, Beran & Brandstätter, Reference Niedermayr, Beran, Brandstätter and Möller1989) or as small accumulations within the hosting gypsum. Gabbroic intrusive and volcanic rocks are common in the western gypsum deposits of the NCA.
The Golling/Moosegg deposit is located in the Juvavic unit of the NCA. The deposit has a poorly constrained age of Late Permian, using stable sulfur isotope data (Pak, Reference Pak1978). The main Haselgebirge body comprises foliated, massive and brecciated anhydrite and gypsum. For the Late Permian period, rifting with a half-graben structure is proposed. In an advanced stage of the rifting gabbroic rocks intruded into a high crustal level; even a few volcanic rocks are known. The volcanic rocks are in close contact with the Haselgebirge; therefore, they are interpreted as syngenetic (Schorn & Neubauer, Reference Schorn and Neubauer2011; Schorn et al. Reference Schorn, Neubauer, Bernroider and Genser2012)
The Wienern/Grundlsee gypsum deposit is situated at the base of the Hallstätter nappe, which is part of the Juvavicum. Some tens of metres of gypsum encase an anhydrite body. A pumpellyite-bearing pillow-lava breccia has been observed at the base of the deposit (Haditsch, Reference Haditsch1968; Kirchner, Reference Kirchner1979).
Lessern and Wildalpen are small gypsum accumulations within the Werfener strata. Geological descriptions are scarce. More than 50 gypsum occurrences are observed only in the eastern part of the NCA (Weber, Reference Weber1997). A gypsum occurrence some kilometres east of Lessern (close to Stainach, Styria) was classified with spores as Late Permian (Klaus & Pak, Reference Klaus and Pak1974).
Tragöß/Haringgraben: The Upper Permian Haselgebirge Formation at Haringgraben represents a c. 100 m thick wedge and hosts one of the largest gypsum/anhydrite deposits of the Eastern Alps. It is stratigraphically situated above the younger, Scythian, Werferner Slates. These units are overlain by a thick carbonate sequence (Wetterstein Formation of Triassic age) (Kölbl & Gawlick, Reference Kölbl and Gawlick1999). Sulfides (sphalerite, galenite, pyrite), sulfarsenides (enargite, baumhauerite) and native sulfur enrichments are known (Postl, Reference Postl, Niedermayr, Brandstätter, Kandutsch, Kirchner, Moser and Postl1990).
2.c. Gypsum deposits of the Central Alpine Mesozoic sampled for this study
The Central Alpine Mesozoic (CAM) hosts a second evaporative zone. Geographically it is situated along with the mostly amphibolite-facies metamorphic crystalline of the Lower Austroalpine nappes from the Semmering area.
Stanz: The gypsum deposit of the Stanz valley is one of the Central Alpine deposits sampled for this study and it lies within a Lower to Upper Triassic unit. The Triassic of the Stanz valley is a few hundreds of metres wide stripe with Scythian quartzites, Anisian rauhwacke and Anisian and Ladinian Muschelkalk. Host rocks of the Triassic series are gneisses, micaschists and quartz-phyllites of the Lower and Middle Austroalpine nappes. The deposit consists of a tens of metres wide anhydrite core, encased by gypsum. The gypsum body is most probably of Carnian or Norian age (Bauer, Reference Bauer1967; Hagenguth, Reference Hagenguth1988).
Göstritztal/Schottwien: Two Central Alpine gypsum deposits are found in the Semmering area: Haidbachgraben and Göstritz. The samples investigated in this study are from the Göstritz valley. Characteristic rocks of the Semmering–Triassic are violet and green Keuper sericite schists with gypsum intercalations. Age data like classification with spores are lacking, but a Carnian age is the most probable. Similar to the Stanz valley deposit, an anhydrite core is enclosed by a gypsum rim. The immediate wall rocks of the Göstritz deposit are Rhaetian dolomites, violet sericite schists, Keuper quartzite and Rhaetian black-shales and limestones (Bauer, Reference Bauer1967).
3. Material and methods
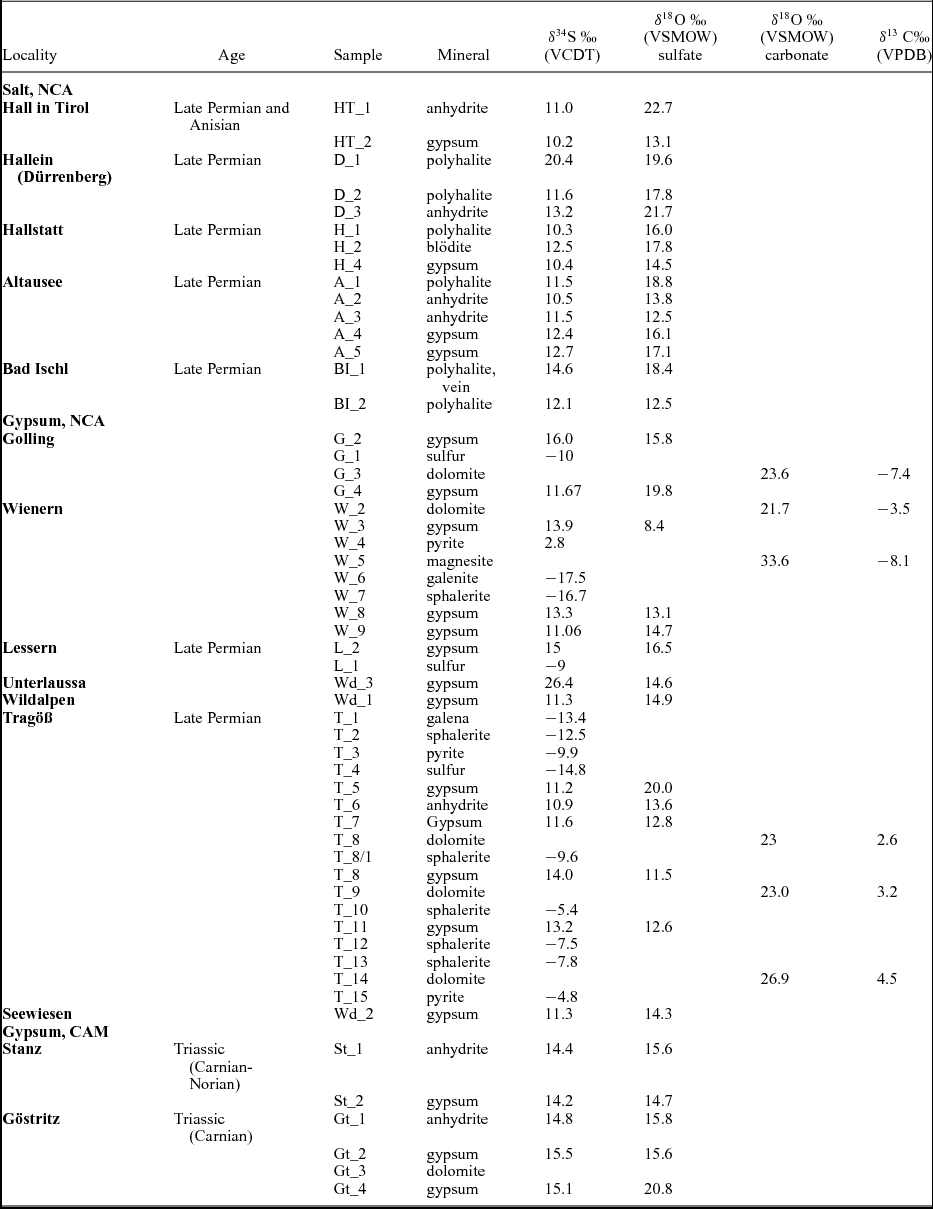
Descriptions of the analysed material, mineral associations, as well as data on stratigraphic age and location are given in the online Supplementary Material available at http://journals.cambridge.org/geo and Tables 1 and 2. Isotopic data on sulfates, sulfides and carbonates are displayed in Table 1. In Table 3 calculated temperatures using sulfur isotope distributions in sulfate, sulfides and sulfur are given.
Table 1. Isotopic composition of sulfate and sulfides from the investigated evaporitic deposits
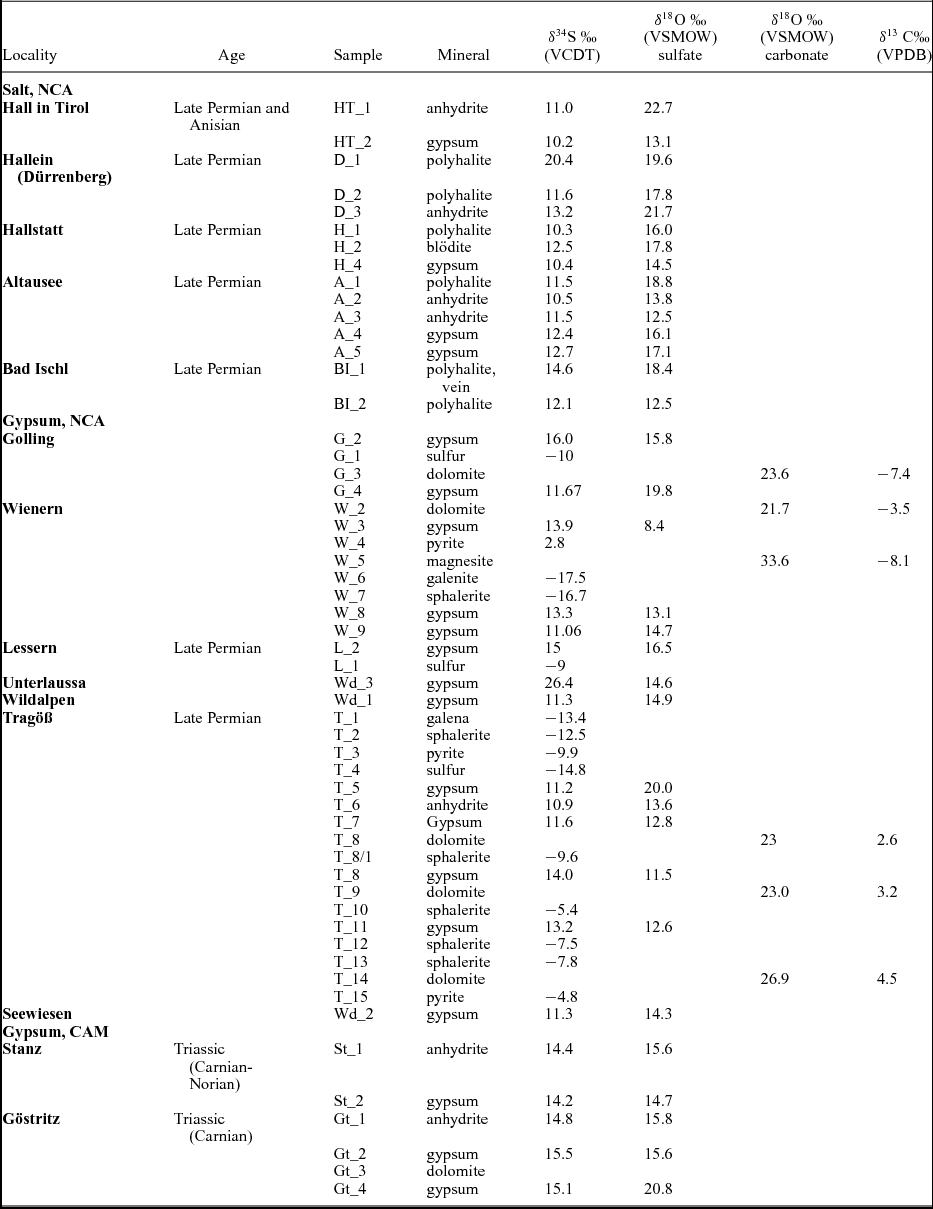
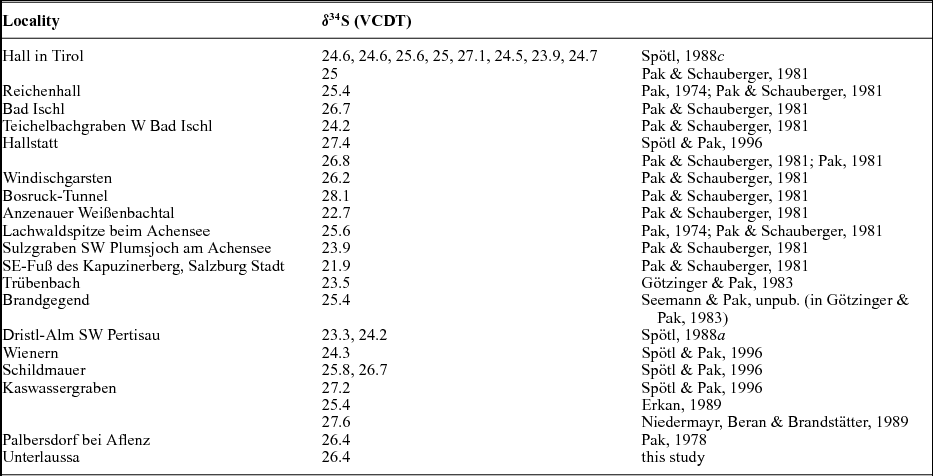
Table 2. Localities of the NCA with ‘Röt-type’ δ34S values
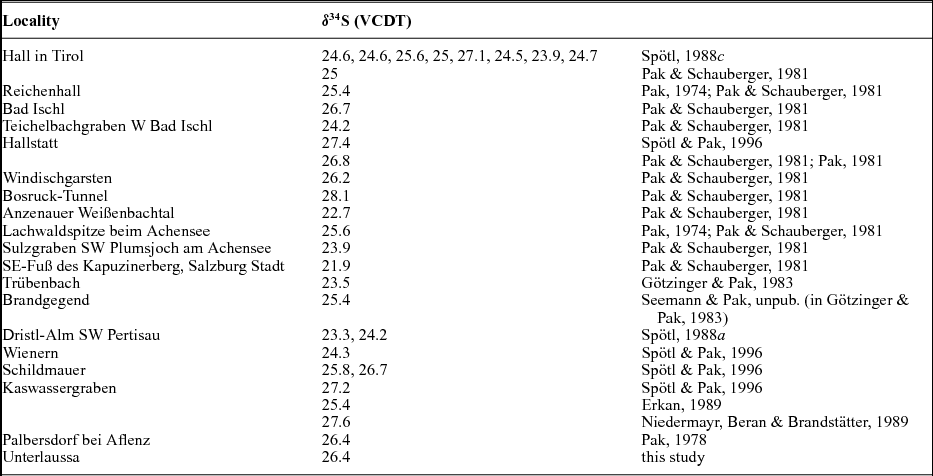
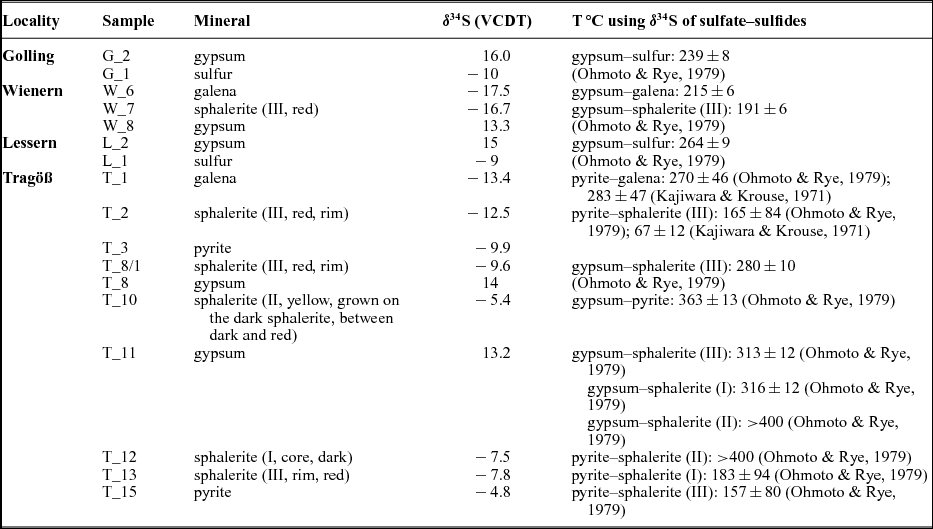
Table 3. Calculated temperatures using sulfur isotope distributions in sulfate, sulfides and sulfur
The isotope ratios of sulfates (δ34S and δ18O) and sulfides (δ34S) were determined by measuring the isotopic composition of resulting SO2 and CO2 gases on a dual-inlet and triple collector mass spectrometer. Sulfur in the form of SO2 gas was quantitatively extracted from BaSO4 samples by thermal decomposition at 850°C in a Cu boat in the presence of Na2PO4 reagent (Hałas & Szaran, Reference Hałas and Szaran2001, Reference Hałas and Szaran2004). CO2 gas was prepared by graphite reduction with conversion of CO to CO2 by glow discharge (Mizutani, Reference Mizutani1971). Nearly quantitative CO to CO2 conversion was attained using a magnetic field in the conversion unit (Hałas et al. Reference Hałas, Szaran, Czarnacki and Tanweer2007). Rough delta values were normalized to the VCDT and the VSMOW scales by analysis of the SO2 and CO2 raw isotopic ratios prepared from the NBS-127 standard, for which we assumed δ34S = 21.17‰ (Hałas & Szaran, Reference Hałas and Szaran2001) and δ18O = 8.73‰ (Hałas et al. Reference Hałas, Szaran, Czarnacki and Tanweer2007).
For carbonate samples, the δ13C and δ18O values were determined as well. CO2 gas was extracted from calcite at 25°C by reaction with H3PO4 (McCrea, Reference McCrea1950) and measured on an isotope-ratio mass spectrometer with dual-inlet system. Standard deviations of measurements for the NBS19 international standard were better than 0.1‰. Delta values were normalized to the Vienna Pee-Dee Belemnite (VPDB). For a rigorous discussion regarding the relationship between SMOW and VSMOW see Sharp (Reference Sharp, Holland and Turekian2014).
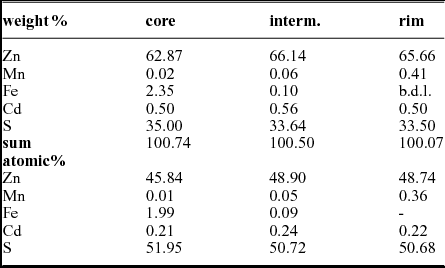
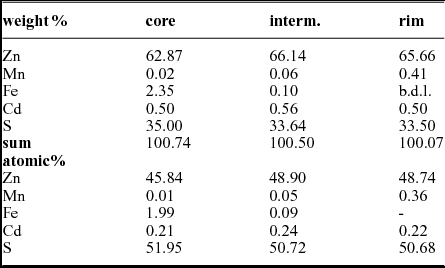
Polished sections of sphalerite where analysed with a Jeol 6610 LV scanning electron microscope equipped with an Oxford WDS 700 spectrometer (20 keV, beam current 20 μA, standards Zn, S: sphalerite, Fe: pyrite, Mn: metallic manganese, Cd: metallic cadmium). The results given in Table 4 are a median of ten analyses each.
Table 4. Electron microbeam analyses of sphalerite zonation from sample T1, Tragöß
b.d.l. – below the detection limit
4. Results
4.a. Stable isotope data
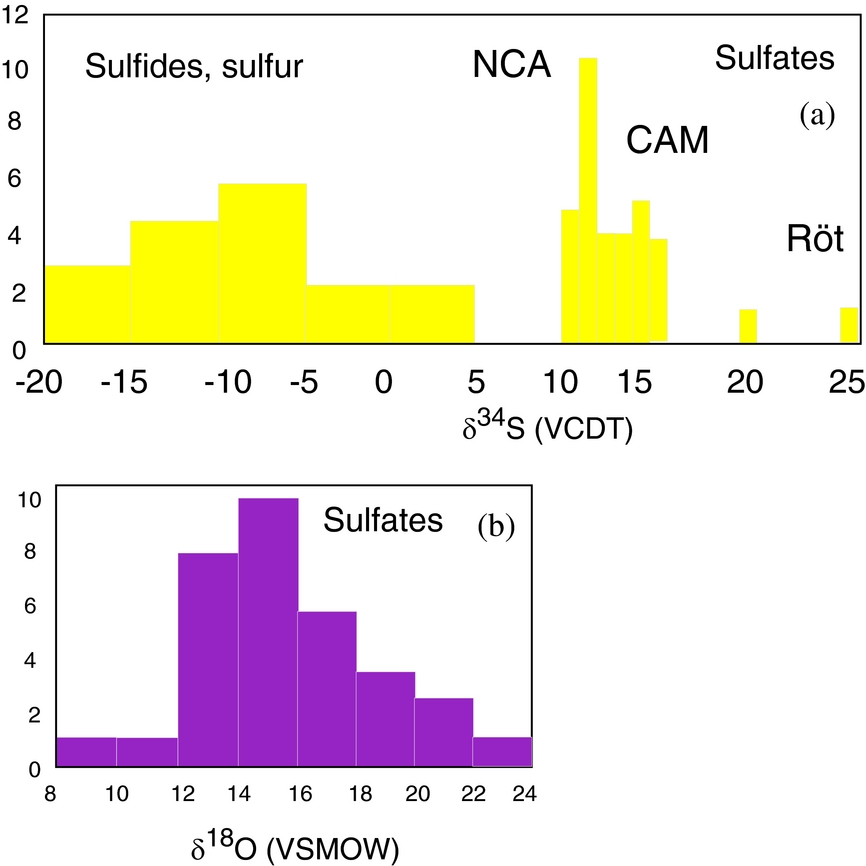
The total number of investigated phases is 54, including analyses on sulfates, sulfides, sulfur and carbonates. The locations of the samples are shown in Figure 1. A total of 34 sulfates were investigated, and oxygen and sulfur isotopic composition were determined for anhydrite, gypsum, polyhalite and langbeinite. The samples containing the investigated phases are described in the online Supplementary Material available at http://journals.cambridge.org/geo and data are given in Table 1. The δ34S values vary between 10.1 and 16‰ (VCDT), with two higher values of 20.5 and 26.6‰. The δ18O values range from 9 to 23‰ (VSMOW) (Figs 2, 3a, b). The δ34S values of 14 sulfides (galenite, sphalerite, pyrite) as well as native sulfur range between −17.5 and 2.8‰ (VCDT). The δ13C and δ18O values were determined for five carbonates as well.
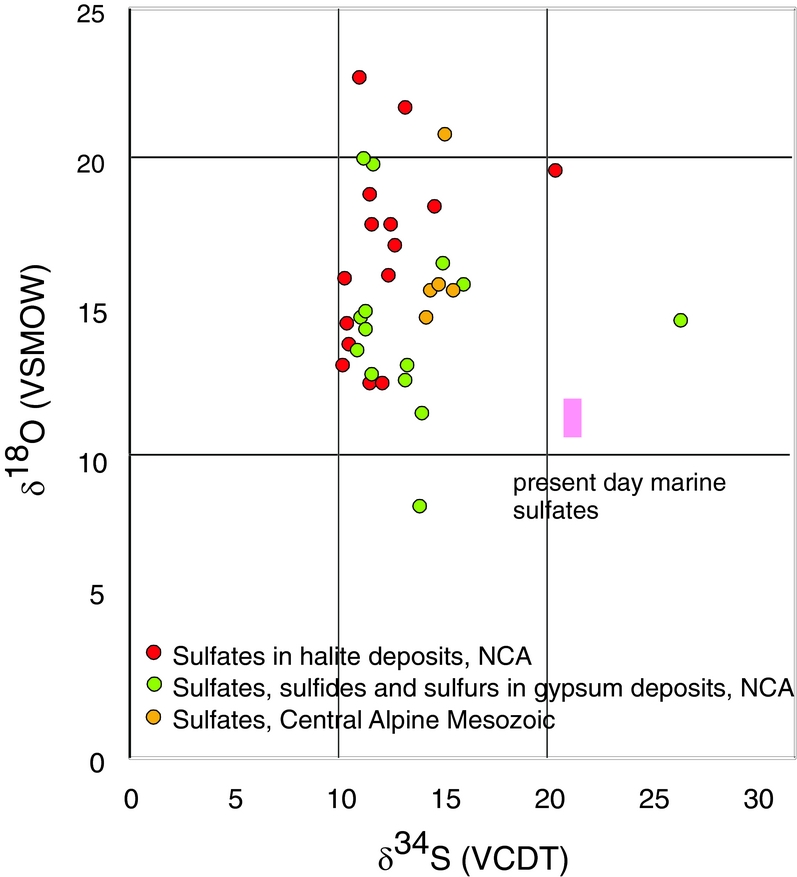
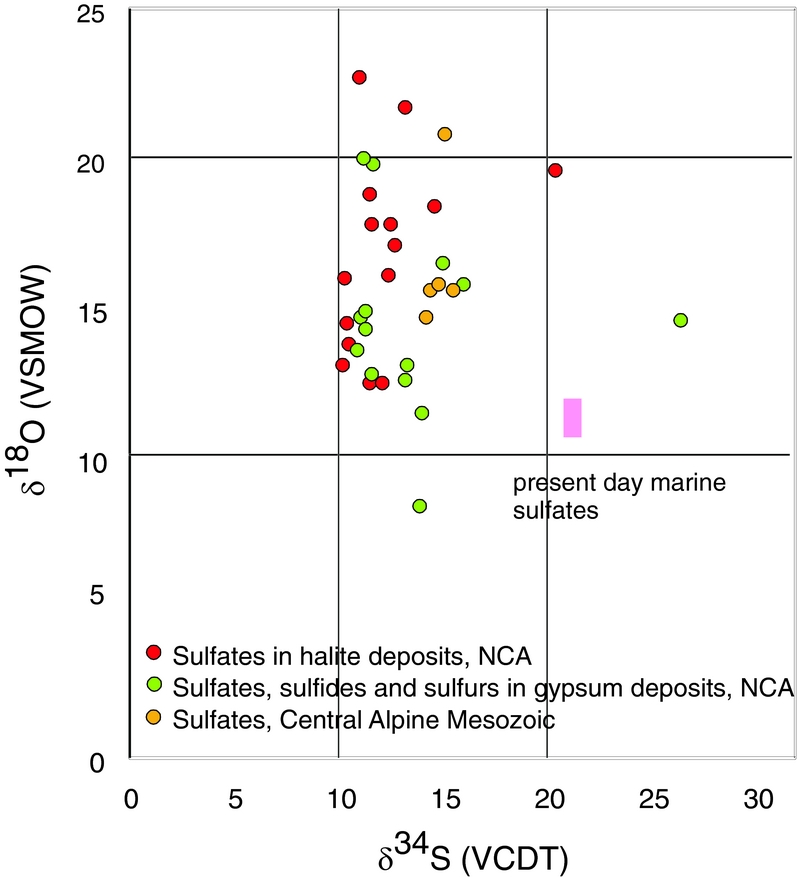
Figure 2. δ34S and δ18O values of measured sulfate. The red circles represent sulfates from the halite deposits; the green circles represent sulfates from gypsum deposits. The rectangle represents the isotope value of sulfate precipitated from marine water.
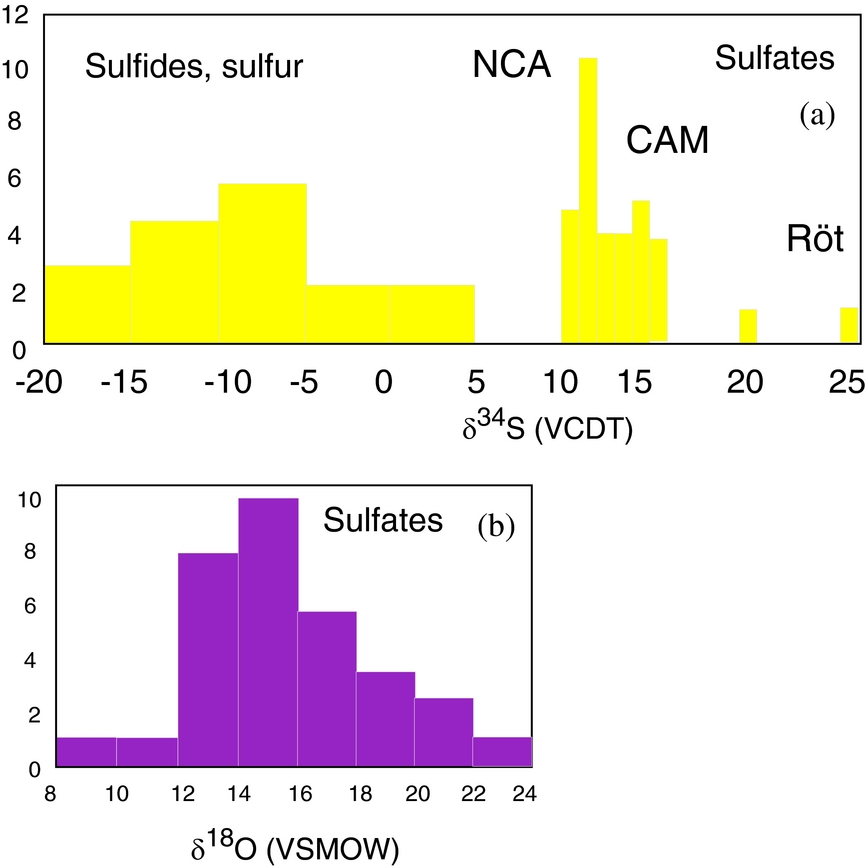
Figure 3. Histograms representing: (a) sulfur isotopic distribution of sulfate, sulfide and sulfur from evaporite deposits; (b) oxygen isotopic distribution of sulfate. All values are measured in this study.
In Figure 2 the isotopic composition of sulfur and oxygen is plotted, differentiated for the different locations of the samples. For comparison, the field covering the present-day isotopic composition for marine sulfates is given as well. The investigated Upper Permian to Triassic sulfates may be divided in three groups with different mean δ34S and δ18O values as follow: (a) from the Permian halite-type deposits of the NCA with mean δ34S and δ18O values of 11.8‰ and 16.6‰, respectively; (b) from the Permian gypsum deposits of the NCA with mean δ34S and δ18O values of 12.6‰ and 14.5‰, respectively; (c) from the Triassic CAM with mean δ34S and δ18O values of 14.8‰ and 16.5‰, respectively.
4.b. Microbeam measurement of sphalerite
In the online Supplementary Material available at http://journals.cambridge.org/geo the measured sphalerite crystal is outlined by a red square. This crystal shows a strong zonation in colour: a brownish core, a yellowish intermediate section and a reddish rim. The results are given in Table 4, representing an average of ten analyses each. The minor component concentrations are low. Cadmium has an average concentration of ~ 0.50 wt%/~ 0.20 at.% irrespective of the position. In contrast manganese and iron show a strong zonation. Iron is concentrated in the core. The average content is 2.35 wt%/1.99 at.%. However, the iron content ranges from 0.4 to 5.88 wt%, with patches of high iron concentrations in a matrix with a lower iron concentration. The core also frequently has small inclusions of pyrite. The manganese concentration is low, close to the detection limit. In contrast the iron content of the outer reddish zone is below the detection limit and the manganese content is enriched compared to the core zone (0.50 wt%/0.36 at.% Mn). The narrow yellowish intermediate zone has low iron (0.10 wt%/0.05 at.%) and low manganese contents (0.06 wt%/0.05 at.%). Other monitored minor elements like (As, Se) were below the detection limit.
5. Discussion
The oxygen isotopic exchange rate is low between dissolved sulfate and water at temperatures below 200°C (Chiba & Sakai, Reference Chiba and Sakai1985; Seal, Alpers & Rye, Reference Seal, Alpers, Rye, Alpers, Jambor and Nordstrom2000). The sulfur and oxygen isotopic composition of sulfates should reflect the composition of dissolved sulfates, and not the composition of water. Besides temperature, the exchange rate is also dependent on pH (Chiba & Sakai, Reference Chiba and Sakai1985).
Dissolved sulfate in modern seawater has a δ34S value of +21‰ (Rees, Junkins & Monster, Reference Rees, Junkins and Monster1978; Longinelli, Reference Longinelli, Fritz and Fonts1983; Böttcher, Brumsack & Dürselen, Reference Böttcher, Brumsack and Dürselen2007), but its composition has varied in ocean history related to bacterial reduction and continental weathering (Claypool et al. Reference Claypool, Holser, Kaplan, Sakai and Zak1980). The δ34S of the seawater sulfate is c. 1.7‰ less than that of the precipitated mineral (Thode & Monster, Reference Thode and Monster1965). In a more detailed study, Szaran, Niezgoda & Hałas (Reference Szaran, Niezgoda and Hałas1998) proposed a value of 1.54±0.09‰. Therefore, sulfur fractionation between sulfate minerals and aqueous sulfates is small, as was underlined by several authors (Ault & Kulp, Reference Ault and Kulp1959; Holser & Kaplan, Reference Holser and Kaplan1966; Sakai, Reference Sakai1968; Raab & Spiro, Reference Raab and Spiro1991). The δ18O value of the present-day dissolved marine sulfate is c. 9.5‰ (Longinelli & Craig, Reference Longinelli and Craig1967; Rafter & Mizutani, Reference Rafter and Mizutani1967; Longinelli, Reference Longinelli, Fritz and Fonts1983), the value of precipitated sulfate being 3.5‰ heavier than the dissolved sulfate, thus c. 13‰ (Gonfiantini & Fontes, Reference Gonfiantini and Fontes1963; Lloyd, Reference Lloyd1968) A more recent study by Szaran, Niezgoda & Hałas (Reference Szaran, Niezgoda and Hałas1998) led to considering an even smaller fractionation value of 2.90±0.09‰.
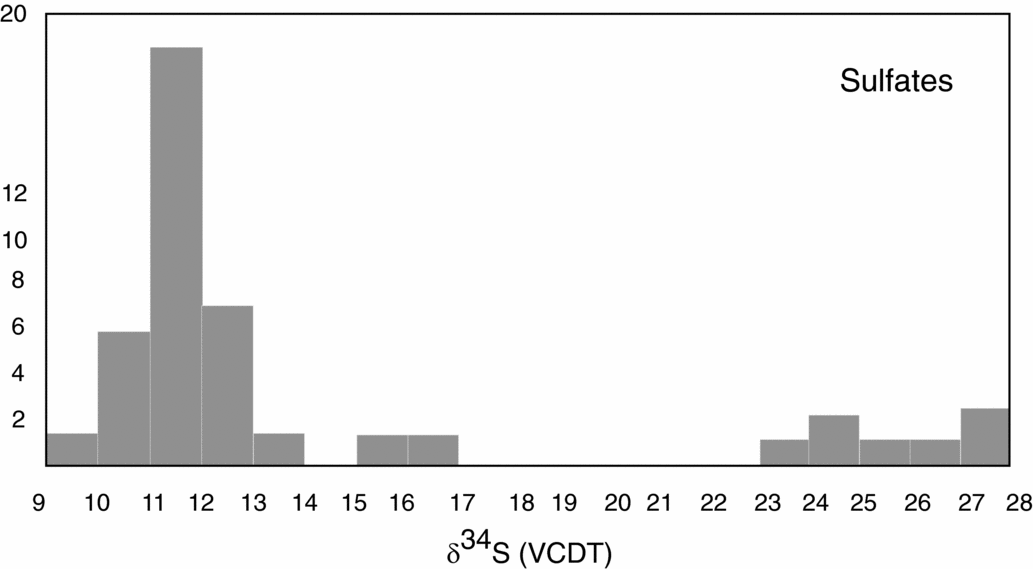
Upper Permian seawater sulfates are characterized by low δ34S values between 10 and 13‰ (Claypool, Reference Claypool, Holser, Kaplan, Sakai and Zak1980). For the Permian, sulfur isotopic composition of the sulfate group from calcitic brachiopod shells shows a higher mean of 13.2±2.5‰ (Kampschulte & Strauss, Reference Kampschulte and Strauss2004). In the present study, the distribution of δ34S values of sulfates from both halite- and gypsum-type deposits of the NCA have values of 11.8‰ and 12.6‰, respectively, in the range determined by Claypool et al. (Reference Claypool, Holser, Kaplan, Sakai and Zak1980) for Late Permian time. For the NCA, there are a few values higher than 13‰, as for example D_1, BI_1, G_2, W_3 and L_2; in all the cases sulfates are associated with vein or vug filling, thus with remobilization. Interestingly, not all the remobilized sulfates (for example H_4 or A_5) are associated with higher values, instead they have values in the range of 10 to 13‰. The measured δ34S values of sulfates for both types of deposits, halite and gypsum from the NCA, show a maxima between 11 and 12‰. For sulfates of the Haselgebirge, Spötl & Pak (Reference Spötl and Pak1996) determined the maxima in the same range (Fig. 4 this study is plotted from table 1 in Spötl & Pak, Reference Spötl and Pak1996). Similar maxima, between 11 and 12‰, were determined for the Upper Permian anhydrite of western Poland (Peryt, Hałas & Hryniv, Reference Peryt, Hałas and Hryniv2010), for Permian Zechstein anhydrites of northern Germany (Kampschulte, Buhl & Strauss, Reference Kampschulte, Buhl and Strauss1998) and for the Upper Permian metamorphosed anhydrites from Italy (Cortecci et al. Reference Cortecci, Reyes, Berti and Casati1981). A larger δ34S range of about 8‰ was observed for Keuper sulfate in northern-central Europe, with δ34S values decreasing on average from Germany to Denmark (Nielsen, Reference Nielsen, Brimblecombe and Lein1989). The δ18O values display a larger scatter from 9 to 23‰, which is even larger than that found for the Upper Permian anhydrites of northern Germany, the Italian Alps or western Poland (Cortecci et al. Reference Cortecci, Reyes, Berti and Casati1981; Kampschulte, Buhl & Strauss, Reference Kampschulte, Buhl and Strauss1998; Longinelli & Flora, Reference Longinelli and Flora2007; Peryt, Hałas & Hryniv, Reference Peryt, Hałas and Hryniv2010). The mean value for δ18O of sulfate from the halite-type deposits is 16.6‰, higher than in the gypsum-type deposits, with a mean of 14.5‰. For the halite-type deposits, there is a correlation between the sulfur and oxygen isotope composition, with the equation of the best fit line y = 1.25x+1.34, R2 = 0.34. This may support that bacterial sulfate reduction affected both sulfur and oxygen isotopic composition of sulfates. Sulfate-reducing bacteria can substantially enhance the oxygen isotope exchange rate between sulfate and water (Fritz et al. Reference Fritz, Basharmal, Drimmie, Ibsen and Qureshi1989; Grinenko & Ustinov, Reference Grinenko and Ustinov1991) and change the isotopic signature of precipitated sulfates. For the gypsum-type deposit no significant correlation between sulfur and oxygen isotopic values could be put in evidence. This suggests that (besides bacterial sulfate reduction) another mechanism was involved for the variable oxygen isotopic values, most likely the oxygen isotope exchange with brine water over the long residence time of the sulfate ions (Hałas & Pluta, Reference Hałas and Pluta2000; Zeebe, Reference Zeebe2010; Boschetti, Reference Boschetti2013). With increasing temperature, isotopic equilibration of oxygen will lower fractionation between sulfate and water, which are in disequilibrium at ambient temperatures. In the present study, the lowest measured oxygen isotope values are c. 12‰, in the range of values indicated by Claypool et al. (Reference Claypool, Holser, Kaplan, Sakai and Zak1980) for the Upper Permian sulfates. Equilibration during thermal overprinting, for example during dehydration of gypsum to anhydrite, will lower the δ18O value of sulfate, assuming a near marine isotopic composition of water. In the present case, as shown in Figure 2, the δ18O values of sulfates show a trend from values of 12‰ towards higher values, up to 23‰. This pattern excludes isotopic re-equilibration at high temperature as an explanation for the observed shift. Instead, the shift towards higher values may be related to recrystallization at relatively low temperatures (30–35°C; Hałas & Pluta, Reference Hałas and Pluta2000).
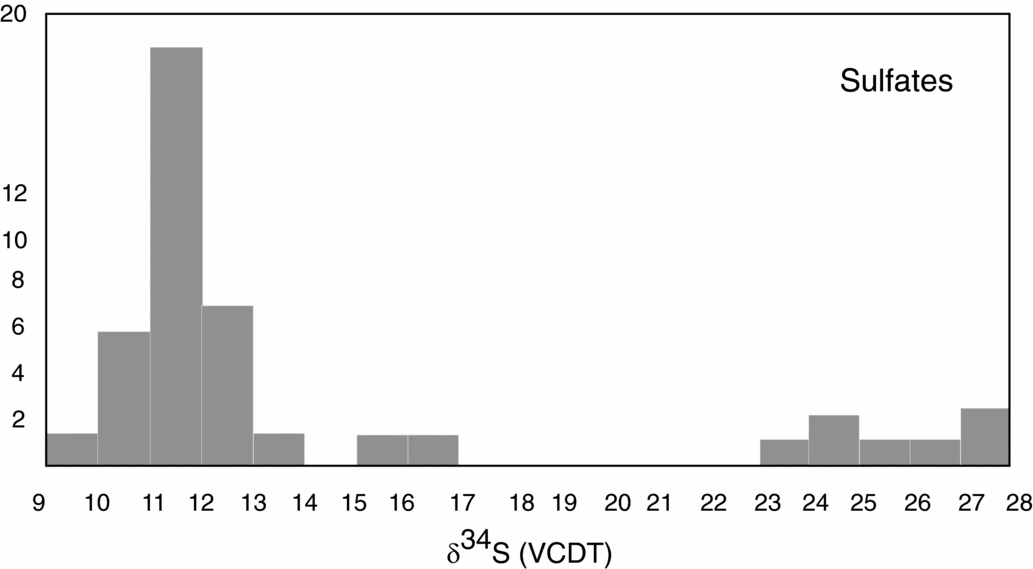
Figure 4. Histogram representing sulfur isotopic distribution for sulfates and sulfides, data after Spötl & Pak (Reference Spötl and Pak1996, their table 1).
The associated carbonates, as calcite, dolomite and magnesite, are in isotopic disequilibrium with sulfates, the isotopic signature of the carbonates indicating rather a primary marine isotopic signature (Table 1) than re-equilibration with sulfates at higher temperatures.
Nielsen (Reference Nielsen1965) mentioned that the so-called Röt-type gypsum of Early Triassic age, measured in deposits of Germany and the Netherlands, have a strikingly high isotopic 34S/32S ratio in comparison to those from the Upper Permian. Nielsen (Reference Nielsen1965) associated such high values with the presence of local, closed basins characterized by high bacterial activity. The sharp increase of the δ34S values from Late Permian to Early Triassic times is considered by Holser (Reference Holser1977) to be ‘the result of . . . fast net sulfide deposition’. In the present study, a gypsum from Unterlausa (Table 1) shows a high δ34S value of 26.4‰. Similar high values were measured by Pak (Reference Pak1974, Reference Pak1978, Reference Pak1981), Pak & Schauberger (Reference Pak and Schauberger1981), Götzinger & Pak (Reference Götzinger and Pak1983), Spötl (Reference Spötl1988 a, c), Erkan (Reference Erkan1989), Niedermayr, Beran & Brandstätter (Reference Niedermayr, Beran, Brandstätter and Möller1989) and Spötl & Pak (Reference Spötl and Pak1996) for evaporites situated in the NCA (Table 2). Our compilation of data for the Röt-type gypsum from the literature (Table 2; Fig. 5) supports the presence of Lower Triassic evaporites, over a larger area than previously estimated. The δ34S values determined for the NCA are lower than those determined for Röt-type evaporites in the Netherlands, Germany and Poland, varying between 27 and 31‰ (Scholle et al. Reference Scholle, Scholle, Peryt and Ulmer Scholle1995). A characteristic feature of the Unterlausa gypsum is the fine-grained texture and the grey colour. X-ray diffraction (XRD) analyses indicate pure gypsum with no visible crystallized detrital components such as quartz or feldspar. According to Kovalevych et al. (Reference Kovalevych, Peryt, Beer, Geluk and Hałas2002) the Röt evaporites were deposited in the E–W-oriented German Basin, which extended from the UK offshore to western Poland. The data compilation displayed in Figure 5 clearly demonstrates the presence of this type of Lower Triassic evaporites also occurring in an E–W trend along the NCA. Recent investigations by Rey et al. (Reference Rey, Amiot, Fourel, Rigaudier, Abdala, Day, Fernandez, Fluteau, France-Lanord, Rubidge, Smith, Viglietti, Zipfel and Lécuyer2016) have shown that land and ocean temperatures strongly fluctuated during Late Permian to Early Triassic times. Moreover, we can correlate the formation of Röt-type evaporites with a temperature maximum during Early Triassic time, as documented by Rey et al. (Reference Rey, Amiot, Fourel, Rigaudier, Abdala, Day, Fernandez, Fluteau, France-Lanord, Rubidge, Smith, Viglietti, Zipfel and Lécuyer2016). From the point of view of ocean chemistry, during the deposition of the Röt evaporites, the concentration of the sulfate ions in seawater was intermediate between the high value, typical for Late Permian seas, and the low value typical for Late Triassic seas (Kovalevych et al. Reference Kovalevych, Peryt, Beer, Geluk and Hałas2002). Worldwide at the level of the Lower Triassic, the perturbation of the carbon cycle and carbon positive excursion were evidenced by Payne et al. (Reference Payne, Lehrmann, Wei, Orchard, Schrag and Knoll2004). The carbon isotopic excursions reflecting major perturbations after the Permian–Triassic extinction event were correlated with ocean anoxia (Song et al. Reference Song, Wignall, Tong, Bond, Song, Lai, Zhang, Wang and Chen2012). In the light of present investigations and available studies, we suggest that Röt-type evaporites were formed in a large intracontinental stratified sea, with limited connection to the ocean during oscillations following the Permian–Triassic mass extinction.
Figure 5. Distribution of Lower Triassic ‘Röt-type’ gypsum (see also Table 3): 1 – Hall in Tirol; 3 – Hallstatt; 5 – Bad Ischl, Teichelbachgraben; 7 – Wienern; 9 – Unterlaussa; 15 – Bad Reichenhall; 16 – Kapuzinerberg Salzburg; 17 – Anzenau Weißenbach; 18 – Pertisau Achensee; 19 – Windischgarsten; 20 – Bosruck-Tunnel; 21 – Schildmauer bei Admont; 22 – Palbersdorf bei Aflenz; 23 – Brandgegend, Trübenbach.
The Upper Triassic evaporites of Carnian–Norian ages from the CAM show a narrow range of δ34S values, close to 15‰. Claypool et al. (Reference Claypool, Holser, Kaplan, Sakai and Zak1980) indicated values between 15‰ and 17‰ from the Middle Triassic to Jurassic. The oxygen isotope composition for the Lower Triassic Röt-type evaporites and for the Upper Triassic evaporites is similar: between 15‰ and 16‰. One exception is represented by the Gt_4 sample, from Göstritz (CAM), a joint fill gypsum, with a δ18O value of 20.8‰ supporting oxygen isotopic exchange with brine water during recrystallization.
When present in visible amounts, sulfides were separated and concentrated. The broad distribution of sulfide δ34S values points towards bacterial reduction of the sulfate group (Berner, Reference Berner1985). During this process, various sulfides, depending on the available cations, were formed. The investigated evaporitic deposits from the NCA were overprinted during burial at higher temperatures (Leitner et al. Reference Leitner, Neubauer, Genser, Borojevic-Sostaric, Rantitsch, Jordan, Mark and Verati2013; Schorn et al. Reference Schorn, Neubauer, Genser and Bernroider2013). We used the sulfate–sulfide and sulfide–sulfide isotopic data in order to calculate the overprint temperatures. The calculated temperatures and associated errors are given in Table 3, for sulfate–sulfide and sulfide–sulfide mineral pairs. The sulfide–sulfide calculated temperatures have larger errors owing to the small isotopic fractionation between various sulfides. For Golling, Wienern and Lessern, the sulfate–sulfide thermometer shows a narrow range of thermal overprint between 215 and 264°C. For Tragöß, the most reliable calculated temperatures, sulfate–sulfide, are higher, between 280 and 360°C. The temperature calculated using the Ohmoto & Rye (Reference Ohmoto, Rye and Barnes1979) calibration and the sulfur isotopic composition of the yellowish sphalerite (II) is considered unrealistically high, reflecting isotopic disequilibrium. For the pyrite–galena mineral pair, the calculated temperatures are similar using either the Kajiwara & Krouse (Reference Kajiwara and Krouse1971) or Ohmoto & Rye (Reference Ohmoto, Rye and Barnes1979) calibrations (Table 3). For the pyrite–sphalerite mineral pair, temperatures are much higher using the Ohmoto & Rye (Reference Ohmoto, Rye and Barnes1979) and lower using the Kajiwara & Krouse (Reference Kajiwara and Krouse1971) calibrations (Table 3). A recent calibration is available for the system pyrite–H2S (Syverson et al. Reference Syverson, Ono, Shanks and Seyfried2015). In this new calibration, the ‘a’ factor of the temperature equation is −0.737 instead +0.4 as given previously in the equation determined by Ohmoto & Rye (Reference Ohmoto, Rye and Barnes1979). No similar recent calibrations are available for other sulfides as sphalerite or galena, so for the moment, the data of Syverson et al. (Reference Syverson, Ono, Shanks and Seyfried2015) could be not applied in calculating mineral pair temperatures. If changes of a similar magnitude and sign for the ‘a’ factor from the temperature equation are expected for the other sulfides, then the calculated temperatures using the mineral pairs will not differ significantly.
Microbeam measurements show a zonation of minor elements in sphalerite (Tragöß, sample T_1, Tables 1, 4). The sphalerite microstructure indicates thermal overprinting, with no microbial structure being preserved. Thermal overprinting is also supported by stable isotope data and temperature calculations. Chemical zoning is related to fluctuating fluid composition or non-equilibrium rather than to a decreasing temperature trend.
6. Conclusions
For the Upper Permian of the NCA, the data indicate that the δ34S isotopic composition of sulfates has a maxima of measured values between 11 and 12‰. The values for the gypsum-type and halite-type deposits are similar, indicating no major fractionation for the different salinity stages of the basin. Bacterial sulfate reduction is supported by the presence of sulfides and sulfur with low and variable δ34S isotopic composition. Sulfates with δ34S higher than 13‰ show remobilization features.
Lower Triassic, Röt-type sulfates are characterized by a heavy sulfur isotopic composition of c. 26‰. The present study supports the fact that these evaporites were widespread over the entire area of the NCA. Data compilation demonstrates that Röt-type evaporites formed in a large intracratonic stratified sea, with limited connectivity to the ocean.
The development of the sulfates of Carnian–Norian age from the CAM is more restricted in areal extent, and sulfates are characterized by δ34S values of c. 15‰.
The mean value for δ18O of sulfate from the halite-type deposit is 16.6‰, higher than in the gypsum-type deposits, with a mean of 14.5‰. Both delta values are higher than the oxygen isotopic composition of sulfates precipitated from present ocean water. δ18O versus δ34S trends support bacterial sulfate reduction and isotope exchange at relatively low temperatures. The oxygen isotope composition for the Lower Triassic Röt-type evaporites and for the Upper Triassic evaporites is similar between c. 15‰ and 16‰.
As a result of thermal overprinting and recrystallisation, bacterial structures are not preserved. However, isotopically, bacterial sulfate reduction was put in evidence. The sulfate–sulfide isotope thermometer indicates overprint temperatures of between 215 and 360°C. Microbeam measurements show several generations of sphalerites related to fluctuating liquid chemistry rather than to variation in temperature.
Acknowledgements
CEEPUS mobility grant CIII-1415-80539 to UMCS Lublin is acknowledged. We appreciate constructive comments on the early version of the manuscript by Christophe Lecuyer and Tadeusz Peryt. Two anonymous reviewers are thanked for careful reading of the manuscript, and valuable comments and suggestions.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/S0016756816000996