


Salmonella enterica is a Gram-negative bacterium that is able to infect a wide range of host species. Infection with S. enterica usually occurs as a result of consuming contaminated food or water, through direct contact with infected animals and by contact with environments contaminated with faecal matter. The nature and severity of disease depends on the Salmonella serotype and the host species involved. There are two main clinical syndromes associated with S. enterica infection: non-typhoidal salmonellosis (a gastrointestinal disease also known as enteritis) and typhoid fever (a systemic disease) (Ref. Reference Layton and Galyov1) (Table 1).
Table 1. Examples of S. enterica serovars, their hosts and disease

S. enterica serovar Typhi (S. Typhi) and S. Paratyphi are restricted to humans and cause systemic diseases (typhoid and paratyphoid fever, respectively). They are important causes of febrile illness in crowded and impoverished populations (with inadequate sanitation) that are exposed to unsafe water and food, and also pose a risk to travellers visiting countries where infection is endemic (Ref. Reference Crump and Mintz2). In 2000, typhoid fever caused an estimated 21.7 million illnesses and 217 000 deaths, and paratyphoid fever caused an estimated 5.4 million illnesses worldwide (Ref. Reference Crump, Lubty and Mintz3). Mortality rates in untreated typhoid fever infections can be 10–15%. In some cases, patients can recover but remain carriers of the bacteria for many years.
S. Typhimurium and S. Enteritidis cause gastroenteritis and nontyphoidal septicaemia (NTS) in humans and in many other animal species worldwide. An important but under-recognised emerging infectious disease problem in sub-Saharan Africa is NTS bacteraemia in children and immunocompromised adults (e.g. AIDS patients). Annually, about 10% of the ~20 million HIV-positive African adults develop invasive NTS infections, resulting in mortality rates above 20% despite antibiotic therapy (Ref. Reference Santos4). S. enterica can cause fatal disease in domestic animals, with severe economic losses, and can persist undetected in food animals, causing a very serious problem for the food industry because of food poisoning.
Current measures to control S. enterica infections are suboptimal. Although antibiotics can kill S. enterica in the blood, whether they can penetrate the Salmonella-containing vacuoles within cells is still questionable (Ref. Reference Lahiri5). Moreover, the emergence of multidrug-resistant strains has reduced the usefulness of most antibiotics. In food manufacturing, prevention by implementation of biosecurity or hygiene measures is expensive and inadequate, and is especially undermined by the expansion of free-range production. Vaccination remains the most feasible means to counteract S. enterica infections; however, currently licensed vaccines for humans and domestic animals are far from optimal, with most based on very old technology and being excessively reactogenic. At present there are no effective vaccines or delivery systems against NTS gastroenteritic or septicaemic infection (Ref. Reference MacLennan6). There is a need to generate new vaccines and antimicrobial strategies, and we argue that understanding within-host population dynamics is important for allowing delivery of targeted intervention.
Infectious diseases can be studied at many levels. Gross parameters, such as host mortality, clinical signs, overall bacterial numbers in the tissue, pathological changes, patterns of shedding, production of immune mediators, and fluctuations and phenotypic profiles of cell populations, have been widely used for descriptive analysis of in vivo infection pathogenesis. Often, we study the gross, cumulative effects of many individual interactions and use some overarching number (e.g. the mean) to represent this. This is only valid if the individual events all operate within the same parameters and distribution. This loses validity if, for example, some apparently identical cells allowed bacterial growth and others did not. Recent advances in molecular and bioimaging techniques have allowed scientists to delve deeper than the ‘mean’ and have enabled studies at the single-cell level in in vivo murine models, leading to an understanding of the location, spread and distribution of individual bacterial populations during infection (Fig. 1).
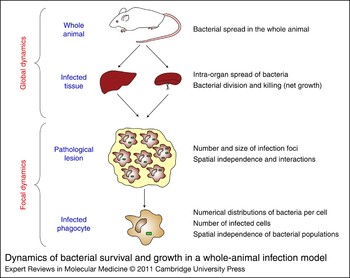
Figure 1. Dynamics of bacterial survival and growth in a whole-animal infection model. Bacterial growth dynamics is studied at different levels of resolution, ranging from the evaluation of parameters in the whole animal, to the study of dynamic interactions of the bacteria with individual pathological tissue lesions, or individual bacterial cells within these lesions.
An understanding of within-host dynamics of S. enterica interactions with eukaryotic cells could shape the design of antibacterial therapies. Specifically, it is important to understand (1) where the bacteria are localised in the various phases of the infection, (2) how the bacteria spread from cell to cell in the whole body, (3) whether there are heterogeneous traits in the behaviour of bacterial subpopulations that might arise during infection as a result of environmental or immunological pressure, and (4) where the bacteria are most vulnerable. Understanding these aspects of the infection is a prerequisite for the development of novel control strategies to (1) target antimicrobials able to act at the right sites of infection; (2) make rational use of combined therapies; (3) to use existing vaccines sensibly; and (4) design new vaccines in a rational way.
This review focuses on recent progress from systemic typhoidal infection of mice in the application of multidisciplinary approaches to study the precise determinants of bacterial growth, spread and distribution at the single-cell level in vivo, and the importance of understanding the dynamics of infection for targeted and rational microbial treatments.
Invasion and spread beyond the gut
S. enterica infects its hosts usually through the oral route. Stomach acid and competition with resident microbial flora constitute early bottlenecks to the infection process. In the distal ileum and caecum, S. enterica invades epithelial cells, and M cells in the Peyer's patches (PPs), using a type III secretion system (T3SS) encoded by genes within Salmonella pathogenicity island 1 (SPI-1) (Ref. Reference Haragga, Ohlson and Miller7) (Fig. 2). Type III secretion systems are a molecular ‘syringe’ that Salmonella (and other bacterial species) use to inject effector proteins through biological membranes of eukaryotic host cells. A subpopulation of hyper-replicating and invasion-primed bacteria has also been identified in the cytoplasm of epithelial cells, and when these cells are extruded out of the epithelial monolayer, they release bacteria that are primed for invasion of the gut (Ref. Reference Knodler8).

Figure 2. Spread of S. enterica in the body during infection.S. enterica invades epithelial and M cells (a) and is engulfed by resident macrophages (b) and dendritic cells (c). Once across the epithelium, nontyphoidal Salmonella strains induce an early local inflammatory response, which results in the infiltration of polymorphonuclear leukocytes (PMNs) (d) into the intestinal lumen and ultimately to diarrhoea. By contrast, Salmonella serotypes that are associated with systemic illness can enter intestinal macrophages and disseminate throughout the reticuloendothelial system (RES) (e) before being captured by resident phagocytes in the spleen and liver (f). Hepatic and splenic multicellular pathological lesions form at the foci of infection.
Subepithelial dendritic cells (DCs) in the PPs take up S. enterica following M-cell invasion (Ref. Reference Hopkins9). Some bacteria can bypass the PPs and disseminate to the spleen and liver directly from the intestinal lumen, being transported by the blood within CD18+ cells (Ref. Reference Vazquez-Torres10). Once they reach the submucosa, some bacteria are captured and killed by resident macrophages. A proportion of the bacteria evades phagocyte killing by the induction of caspase-1-mediated cell death of resident macrophages mediated largely by the SPI-1-encoded T3SS (Ref. Reference Hersh11). The role of flagellin in the induction of caspase-1-mediated cell death in vivo remains controversial (Refs Reference Winter12, Reference Miao13). This process also leads to cleavage of the precursors of IL1β and IL18 to produce bioactive inflammatory cytokines that mediate the recruitment of polymorphonuclear phagocytes (PMNs) and initiate gut inflammation (Ref. Reference Raupach14). Products of the S. enterica SPI-1 sipA and sopABDE2 genes also contribute to inflammation in this phase of the infection by eliciting the production of GROα, GROγ, IL8 and GCP-2 chemokines (Ref. Reference Zhang15). Invasion and gut inflammation have a negative effect on resident microflora, and S. enterica triggers and exploits inflammation to compete with the intestinal microbiota (Ref. Reference Stecher16). The actual role of macrophage death and inflammation in the spread of S. enterica is still unclear (Ref. Reference Guiney17).
S. enterica can also penetrate the gut wall using an SPI-1-independent mechanism, whereby bacteria are engulfed by DCs that open the tight junctions between epithelial cells and might be transported to distant sites (Refs Reference Vazquez-Torres10, Reference Rescigno18). Other cell populations, such as B cells and innate secretory antibodies, also contribute to the control of the spread of S. enterica beyond the gut, as suggested by the lower LD50 value for IgM −/− mice after primary oral infection with wild-type bacteria (Ref. Reference Mittrucker19) and by the increased susceptibility to infection of polymeric immunoglobulin receptor (Pigr −/–) knockout mice, which are unable to bind and actively transport dimeric IgA and pentameric IgM to the mucosae (Ref. Reference Wijburg20).
From the gut to the systemic compartment: S. enterica reaches an intracellular location in the spleen and liver
After the invasion process is completed, Salmonella serotypes that are associated with systemic illness spread from the gastrointestinal tract to the bloodstream, with a proportion of the bacteria passing through the mesenteric lymph nodes (MLNs). MLNs represent a restrictive site for growth and dissemination of S. enterica, because mesenteric lymphadenectomised mice experience accelerated colonisation of the spleen and liver and increased mortality (Ref. Reference Voedisch21). In the blood, the bacteria can be found either extracellularly (where they are opsonised but not lysed by complement) or associated with CD18+ leukocytes (Refs Reference Vazquez-Torres10, Reference Warren22). From the blood, S. enterica reaches an intracellular location within phagocytes of the liver, spleen and bone marrow (Refs Reference Biozzi23, Reference Dunlap24). Early in infection, in the spleen, red pulp macrophages (F4/80+, MSR-Alow) and marginal zone macrophages (MSR-A+) appear to contain most of the bacteria (Ref. Reference Salcedo25). In the liver, S. enterica localises preferentially in resident Kupffer cells. A proportion of bacteria is also found within PMNs (Ref. Reference Richter-Dahlfors26). Only occasionally are bacteria found in DCs or B cells (Refs Reference Yrlid27, Reference Tam28).
Bacterial distribution and survival in the early stages of infection
After arrival of the bacteria in the spleen and liver, the initial foci of infection consist of spatially separated phagocytes, each of which contains a single bacterium (Ref. Reference Sheppard29) (Fig. 3). Early in infection, a reduction in the total number of viable bacteria is seen in the organs of infected mice (Ref. Reference Grant30). This is due to the antimicrobial action of host phagocytes through reactive oxygen species (ROS) generated by NADPH oxidase (Refs Reference Grant30, Reference Mastroeni31, Reference Vazquez-Torres32). Binding of tumour necrosis factor alpha (TNFα) to the receptor TNFR55 facilitates the localisation of vesicles containing NADPH oxidase to vacuoles containing S. enterica, whereas genes contained within Salmonella pathogenicity island 2 (SPI-2) tend to counteract this process (Ref. Reference Vazquez-Torres33).

Figure 3. Population dynamics of S. enterica in the organs of infected mice. Bacteria infect resting phagocytes (a). The infection begins with one bacterium per cell (b) and an excess of uninfected cells (not shown). Inflammatory phagocytes are recruited to the sites of infection with the formation of multicellular pathological lesions that trap the bacteria within discrete foci (c). Host cells within the lesions become activated by immune signals (d). Growth of virulent S. Typhimurium in vivo is also associated with escape from infected macrophages and dissemination to other uninfected cells (e). This is a necessary step for the overall net growth of bacteria in the organs, and results in the formation of new infection foci (e, f). With the progression of infection, each discrete focus becomes a multicellular pathological lesion surrounded by normal tissue, and initially consisting of polymorphonuclear cells (PMNs) with an increasing proportion of inflammatory mononuclear cells over time (f). BMD, bone marrow derived.
The true dynamic nature of events occurring during the very early phase of the disease has recently been captured through population analysis of wild-type isogenic-tagged strains (WITS). Each of these carry a different DNA signature tag in the same noncoding region of the chromosome, allowing quantification of each clone by quantitative PCR combined with metapopulation mathematical models. This approach has shown that both NADPH-oxidase-dependent bacterial death and concomitant rapid bacterial replication occur early in infection, and lead to the establishment and amplification of independent subpopulations of bacteria in tissues (Ref. Reference Grant30).
Bacterial growth and formation of multicellular pathological lesions
Many S. enterica host defence mechanisms and bacterial factors are known to affect net growth rates in the host. The initial reduction in bacterial number is followed by a phase of bacterial net growth underpinned mainly by bacterial division, with microbial death becoming negligible (Refs Reference Grant30, Reference Hormaeche34). The rate of bacterial division is dependent on the virulence of a given S. enterica strain and is controlled by both ROS (Refs 30, 31, 32) and the Slc11a1 gene (Refs Reference Hormaeche35, Reference Hormaeche36, Reference Hormaeche37), which encodes a phosphoglycoprotein of 90–100 kDa that is preferentially localised in membranes of phagosomes containing bacteria and functions as a divalent metal (Fe2+, Zn2+, Mn2+) ion pump at the membrane of the late endosome. Slc11a1 has two variant alleles and the resistance allele is dominant. Susceptibility is associated with a substitution at residue 169, where glycine is replaced by aspartic acid (Refs Reference Blackwell38, Reference Forbes and Gros39).
Analysis of S. Typhimurium WITS population dynamics within and between organs of mice has shown that, in the first 24 h of bacterial growth in a systemic infection, different S. Typhimurium subpopulations escape from infected cells and spread within each organ with no detectable bacteraemia or mixing of subpopulations between the spleen and liver (Ref. Reference Grant30). This is intriguing given the homogeneous distribution of different subpopulations within an organ and the fact that the increase in the number of infection foci is likely to require lysis of infected cells and release of the bacteria in the extracellular compartment (Refs Reference Sheppard29, Reference Grant30, Reference Brown40). Later in the infection, the bacteria continue to divide and the infection enters a truly systemic phase with the onset of bacteraemia and the mixing of subpopulations between organs (Ref. Reference Grant30).
During the bacterial growth phase, inflammatory phagocytes are recruited to the sites of infection, and multicellular pathological lesions are formed that trap the bacteria within discrete foci. PMNs are the first inflammatory cells to infiltrate the foci of infection to form pathological lesions and have a role in host resistance in the early stages of infection (Refs Reference Richter-Dahlfors26, Reference Conlan41). Mononuclear cells that migrate into the organs gradually replace PMNs in the pathological lesions, and in some host–pathogen combinations determine suppression of bacterial growth and survival of the host, and eventually allow the T-cell-dependent clearance of pathogens from infected tissues. The formation of these pathological lesions is a dynamic process that requires an influx of cells from the bone marrow (Ref. Reference Hormaeche42), the presence of adhesion molecules such as ICAM1 (intercellular adhesion molecule 1) (Ref. Reference Clare43) and the balanced action of cytokines. TNFα is the key mediator of cell recruitment into the lesions (Ref. Reference Mastroeni, Skepper and Hormaeche44). IFNγ produced by NK cells and T cells in response to IL12 and IL18 mediates activation of the recruited cells and enhancement of their antimicrobial functions, which in this phase of infection are mainly due to the production of reactive nitrogen species (Refs Reference Mastroeni31, Reference Vazquez-Torres32). Failure to form pathological lesions or to activate cells within the lesions results in abnormal growth and dissemination of the bacteria in infected tissues (Refs Reference Mastroeni, Skepper and Hormaeche44, Reference Everest, Roberts and Dougan45, Reference Mastroeni46).
Bacterial spread in tissues: dispersive infections with low bacterial numbers per cell
One of the key features of the spread and distribution of S. enterica in tissues is the fact that intracellular bacterial densities are heavily skewed towards low intracellular numbers (Refs Reference Sheppard29, Reference Brown40, Reference Grant47, Reference Grant48). In host–pathogen interactions where the bacteria grow at different rates, the increase in the number of infected cells parallels the bacterial net growth rate (Refs Reference Sheppard29, Reference Grant48). The same applies to the multicellular pathological lesions whose numbers also increase in parallel with the microbial burden, resulting in a small increase in the size of the lesion (Ref. Reference Sheppard29).
There are several possible explanations for the low intracellular densities seen in infected tissues during a systemic S. enterica infection. It is possible that death and lysis of the infected cell occur when low bacterial numbers are reached, and necrotic cell death in infected cells has been shown to occur (Ref. Reference Guiney17). It is also possible that only a minority of infected host cells is permissive to bacterial replication and the majority of phagocytes is efficient at restraining the division of the microorganism. This scenario would be plausible, given the difficulty of documenting intracellular replication in mononuclear cells in vivo (Ref. Reference Hsu49), the known vertical (differentiation stage) and horizontal (anatomical source) heterogeneity within phagocyte populations (Ref. Reference Gordon and Taylor50), and the recent identification of nonreplicating dormant intracellular populations of S. enterica within cells (Ref. Reference Helaine51). The dynamic nature of the quantitative basis of S. enterica dispersiveness is, however, difficult to resolve by simple analysis of biological data (Fig. 4). A combination of the distributional data obtained from the direct observation of bacterial populations in tissues of mice and mathematical models has indicated that it is not necessary to invoke the above scenarios as the main determinants of skewed intracellular distributions (and therefore the high level of dispersiveness of S. enterica infections) (Ref. Reference Brown40). Mathematical models that predict a constant rate of stochastic cell lysis in infected phagocytes, independently of intracellular bacterial numbers, can fully capture the observed distributional pattern of S. enterica in host cells in vivo (Ref. Reference Brown40). In other words, the models predict that the majority of bacterial cells replicates within phagocytes, and host cells containing high bacterial numbers would have the same probability of undergoing lysis as cells containing one or a few bacteria. The models also predict density-dependent progressive reductions in intracellular bacterial division rates, indicating that salmonellae would replicate faster in those cells that contain lower bacterial numbers (Ref. Reference Brown40).

Figure 4. Towards a systems-based approach to study S. enterica infections. Iterative feedback between biological research and mathematical modelling has recently proved to be a very powerful approach to capture key aspects of S. enterica spread and distribution in vivo. Experimental results can inform the biological models directly (a). Mathematical models and statistics are fitted to results for increased resolution (b). Predictions of mechanistic bases of biological scenarios, which are difficult to resolve by simple analysis of data, help refine the biological model (c). Predictions from mathematical models can be tested in targeted experiments (d). In this process, each piece of data contributes to validating and improving the models and each prediction can be tested experimentally.
Overall, this indicates that the bacteria grow within discrete infection foci within an organ, escape from these sites to disseminate throughout that organ, and establish new foci of infection at distant sites. Simultaneous infections with different bacterial populations that can be distinguished by fluorescence markers or immunostaining have shown that each focus of infection contains a single bacterial population, and therefore indicates that each new focus of infection originates from the clonal expansion of an individual founder bacterium (Ref. Reference Sheppard29).
The formation of new infection foci is likely to represent a ‘hit-and-run’ strategy where the bacteria spread from cell to cell and from lesion to lesion to stay one step ahead of the local escalation of the immune response that progressively occurs at the level of each infection focus, and to avoid the spatial and nutritional constraint that could result from high intracellular densities (Fig. 3). This would indicate that, contrary to what is suggested by in vitro studies, the ability to grow inside the majority of infected phagocytes and avoidance of intracellular killing (Refs Reference Fields52, Reference Harrington and Hormaeche53, Reference Lissner, Swanson and O'Brien54) are no longer the only prerequisites for S. enterica virulence. The dispersive nature of the infection determined by escape from infected cells, and spread in the tissues are also required for expansion of the initial infecting dose.
Variables that affect the spread and distribution of S. enterica in the body
To fully capture the pathogenesis of S. enterica systemic infections, it is important to establish which host and pathogen variables determine the spread and dispersiveness of the infection. Intrinsic to the constant rate of stochastic cell lysis predicted by current mathematical models of S. enterica infection is the correlation between intracellular densities and bacterial division rates (Ref. Reference Brown40). The models predict higher intracellular bacterial densities in those situations where bacterial division is accelerated (Ref. Reference Brown40). This correlation between bacterial growth and distributional bias can be understood as a consequence of an increase in the relative exposure to host-cell lysis in those infections where the bacteria grow more slowly. Under these conditions, the intracellular bacterial load takes longer to reach a given size, so the probability of lysis before reaching that size will be greater.
The predicted correlation between bacterial growth rates and intracellular bacterial densities has been tested using two approaches. Firstly, infections were performed in immunocompromised animals where enhanced overall net bacterial growth and increased division occur. Higher intracellular densities were observed in the spleens and livers of IFNγ-, TLR4- and gp91phox-knockout mice, confirming predictions of the model (Ref. Reference Grant48). A dichotomy therefore appears in how immunological pressure shapes the patterns of bacterial spread in salmonellosis. In fact, some components of the host immune response (e.g. ICAM1, TNFα) mediate the confinement of bacteria to discrete, localised, infection foci through the formation of multicellular pathological lesions. However, elements of the immune response that mediate activation (loss of IFNγ and TLR4) or expression of the antimicrobial functions (production of ROS) of individual phagocytes, despite slowing down the overall bacterial growth process, also force dispersiveness of the infection at the single-cell level. Second, it was observed that intracellular bacterial numbers per cell are higher when performing infections with S. enterica strains of increasing virulence (Ref. Reference Sheppard29), thus confirming the predicted correlation between bacterial growth rates and intracellular densities.
The predicted density-dependent slowing in intracellular bacterial growth suggests that one of the requirements for S. enterica to grow efficiently is the escape of bacteria from heavily infected cells (presumably by inducing necrotic lysis of the host cell) to infect new phagocytes, thus lowering the intravacuolar bacterial burden.
Is lysis of infected cells the only mechanism of S. enterica spread?
The dispersiveness of infection is underpinned by a branching process, with an increase in the number of foci that largely parallels overall escalation in the bacterial load. During this process, infected cells most likely undergo necrotic lysis that would release individual bacteria in to extracellular space. However, it is possible and probable that alternative methods of bacterial cell spreading exist during salmonellosis. For example, liver phagocytes can undergo apoptotic caspase-3-mediated cell death in vivo, with apoptosis being more frequent in heavily infected cells and a proportion being dependent on the presence of TLR4 (Ref. Reference Grant47). Cell-to-cell spread by the induction of apoptosis in host cells would provide a mechanism by which the bacteria could spread without being exposed to the extracellular environment. In fact, apoptotic cells with intact membranes are engulfed by phagocytes in vivo, and therefore it is reasonable to assume that infected apoptotic host cells would die without releasing their load of bacteria extracellularly, thus allowing S. enterica potentially carrying several bacteria to spread intracellularly.
Bacterial spread through apoptotic bodies might appear incompatible with current mathematical and biological models for the spread and distribution of S. enterica in mammalian tissues (Ref. Reference Brown40) where extracellular release of individual bacteria following cell lysis is assumed to be the basis of the observed increase in the number of infected cells during infection. However, careful analysis illustrates that caspase-3-mediated apoptosis is a rare event, and phagocytosis of infected apoptotic bodies would lead, within the model, to a situation where a new infected host cell simply replaces the previous host cell containing the same set of bacteria, allowing neither the redistribution of single bacteria nor an increase in the number of foci of infection without violating the model assumptions (Ref. Reference Grant47). There is therefore evidence for two distinct mechanisms of S. enterica-induced phagocyte death operating in parallel, but governed by different dynamics (Fig. 5). Caspase-3-mediated cell death is rare and correlates with intracellular bacterial density, whereas the more common necrotic lysis and extracellular release of individual bacteria are independent of intracellular bacterial numbers (Ref. Reference Grant47).

Figure 5. Factors that control the intracellular and extracellular phases of infection.S. enterica grows in phagocytic cells (a). Cell-mediated immunity enhances the antibacterial functions of phagocytes and probably also contributes to host-cell lysis (b). The growth of virulent S. Typhimurium in vivo is also associated with escape from infected macrophages and dissemination to other uninfected cells. This is a necessary step for the overall net growth of bacteria in the organs, and results in the formation of new infection foci and is likely to occur mainly as a consequence of necrosis of infected cells and establishment of new foci of infection by released bacteria (c). During their cell-to-cell spread, bacteria are transiently present in extracellular space, where they are opsonised by antibodies and targeted to Fc receptors (FcR) (d). This process enhances bacterial internalisation by macrophages through FcγRI (e) and activates cellular antibacterial functions dependent on the production of reactive oxygen intermediates. We have also observed that S. enterica can cause caspase-3-dependent classical apoptosis of infected cells in the liver (f), thus opening the possibility that an alternative mechanism exists that would allow the bacteria to spread from cell to cell without being released extracellularly (g).
Clinical implications
Successful medical intervention or vaccine-induced prevention against bacterial pathogens such as S. enterica would need to satisfy the following key requirements: (1) abort or reverse rapidly the overall numerical escalation of bacterial load in tissues; (2) halt the spread of bacteria in the body; and (3) rapidly clear the bacteria from sites of infection, leading to elimination of pathogens. This will require targeting of appropriate drugs or immune responses towards suitable anatomical locations at the right point during the infection. For example, some drugs (e.g. gentamicin) or immune responses (e.g. antibodies) can only target bacteria in the extracellular space, whereas others can also target bacteria in the intracellular compartment (e.g. antibiotics that penetrate cells and cell-mediated immunity). The determinants of the net growth rate of bacteria in tissues also need to be understood before considering treatment, because some antimicrobials preferentially kill bacteria that are replicating, whereas other drugs are also capable of attacking nondividing microorganisms. Recently, evidence for dormant bacteria within cells has corroborated the importance of understanding the true nature of bacterial growth rates in vivo (Ref. Reference Helaine51).
A thorough understanding of the mechanisms by which S. enterica spread within tissues and between organs could have profound implications for rational choices in prevention and treatment. Intracellular (e.g. apoptosis-mediated) and extracellular (mediated by necrotic lysis) routes of bacterial spread in tissues can also have vastly different consequences on bacterial susceptibility to established immune responses (e.g. humoral versus cell-mediated immunity) or antimicrobial intervention strategies. Therefore, the ability to bet-hedge (Refs Reference Jansen and Stumpf56, Reference Kussell and Leibler57) by mixing these two strategies of cell-to-cell transfer might broaden the persistence of S. enterica in the face of an ever-changing host environment. Indeed, bacterial persistence in apoptotic bodies might explain why antibiotics that poorly penetrate host cells (e.g. gentamicin) fail to completely clear all bacteria during an infection (Ref. Reference Bonina, Costa and Mastroeni58). The possibility that bacterial spread might come to a halt in the later phases of sublethal infections, where there is no increase in bacterial numbers (and therefore no increase in the number of infection foci), might explain the difficulty in clearing pathogens from the tissues using antimicrobial treatments based on β-lactam antibiotics that are efficacious on bacteria that actively replicate (Ref. Reference Maskell and Hormaeche59). The existence of extracellular bacterial spread (following necrosis) and intracellular spread (following apoptosis) provides a rationale for the requirement for both antibodies and T cells in immunity to S. enterica and for the limited efficacy of those S. enterica vaccines that elicit only one of the above-mentioned branches of the immune response.
The combination of biological research on the localisation, spread and distribution of S. enterica with mathematical models of infection can be used as a novel approach to generate testable predictions to evaluate the probable impact of new treatments or immune control on the fate of infection. Using control contours based on current branching process mathematical models of S. enterica infection, it is possible to predict, in various host–pathogen combinations, the levels of intra- and extracellular control needed to achieve clearance of the microbial population from tissues (Ref. Reference Brown40). Useful predictions can also be made for combined antimicrobial treatments. For example, current models predict that simply slowing down intracellular bacterial growth (e.g. with bacteriostatic drugs or immune mechanisms such as RNI) would enhance the efficacy of interventions that act on extracellular bacteria. The prediction of density-dependent slowing in intracellular division of the bacteria suggests that factors, or interventions, causing a delay in host-cell lysis would be particularly beneficial in slowing down the spread of an infection, by decreasing the mean rate of intracellular proliferation.
Research in progress and outstanding research questions
Central to a complete understanding of any disease is the ability to integrate information at different scales into a coherent model that fully explains the disease process. One challenge that remains is how to move from our ever more detailed understanding of cellular and molecular microbiology in artificial laboratory systems towards an explanation of the dynamics of pathogen survival and growth in a whole-animal infection model. Historically, the effect of prevention or treatment on infection has been studied largely by compartmentalised approaches assessing gross parameters such as host mortality, clinical signs and overall bacterial numbers in tissues and correlating these with the presence, absence (or experimental ablation) and phenotypic characterisation of immune responses. Although these studies have demonstrated associations between the control of pathogen growth and activity of antimicrobials or specific components of the vaccine-induced response, they often fall short of identifying the precise mechanisms underlying these associations. Such mechanisms could potentially be dependent on a very large number of variables that cannot always be comprehensively gathered in vivo.
In relation to S. enterica infections and this review, bacterial growth, death and spread in the body are still undetermined aspects of the infection process despite their relevance to vaccination and treatment. The ongoing challenge is to develop an understanding of the dynamic interactions between host and bacterial mechanisms that determine the net growth rates of S. enterica subpopulations within the host. The aim will be to use integrated approaches where advanced molecular, proteomic and microscopy techniques, in combination with mathematical and statistical models, will enable a systems-level in vivo analysis of S. enterica–host interactions, to underpin rational translational research into the development of vaccines and immunotherapeutics. Specific goals should include: (1) determining how salmonellae exit host cells in the systemic compartment and how this affects redistribution within the host and the establishment of new foci; (2) determining the dynamics of S. enterica adaptation to the in vivo environment; and (3) determining how different classes of antibiotics and vaccine strategies affect bacterial distribution and within-host dynamics.
Determining how salmonellae exit host cells in the systemic compartment
The use of techniques such as WITS, fluorescence microscopy and two-photon intravital imaging to study individually infected cells will give unprecedented fine-structure data concerning within-host S. enterica infection dynamics. Monitoring the qualitative and quantitative population structure of bacteria recovered from different anatomical locations together with the modelling of data will demonstrate ways in which different components of the innate and acquired immune system control the expansion (net growth rate) and determine the contraction (clearance) of the bacterial load in tissues. These studies will provide a new perspective on the events involved in S. enterica containment and escape, and will provide substantial refinement of our understanding of the variables that govern the dynamics of how S. enterica interacts with and spreads within tissues, which is important for the continued development of novel therapeutic strategies.
Determining the dynamics of S. enterica adaptation to the in vivo environment
As infection progresses, the bacteria are exposed to an increasingly hostile environment as a result of the progressive escalation of a complex host immune response. Consequently, to continue surviving and growing within tissue phagocytes, S. enterica is likely to require the coordinated and sequential regulation of fitness genes. Bacterial gene regulation has so far been investigated largely using exposure to artificial environmental conditions or to in vitro cultured cells, and little information is available on how S. enterica adapts in vivo to sustain cell division and survival during the various phases of the infection. Identification of the S. enterica genes and proteins that are regulated during infection will help in understanding the molecular mechanisms that control the host–pathogen interaction that could provide new targets to disable transmission with preventative or therapeutic interventions.
Determining how different classes of antibiotics and vaccine strategies affect bacterial distribution
Most interventions to control bacterial infections rely on antibiotics and vaccines, but little is known about how these work in vivo. Rather, inferences are made from in vitro systems, but rarely are these ideas tested properly in an in vivo infection model. Many key points concerning antibiotic therapy remain to be addressed. These include: (1) why antibiotic regimens fail to completely clear an infection; (2) how surviving bacteria evade antibiotics; and (3) to what extent the immune system and consequent dynamics of bacterial growth and persistence contribute to these phenomena. A clearer understanding of how antibiotic treatments affect bacteria at the level of infected cells, and the distribution of bacteria in infection foci, has implications for targeting antimicrobials to appropriate sites of infection.
Similarly, we still poorly understand how the infection evolves in the presence of vaccine-induced immunological pressure. Some key outstanding questions remain: (1) Is there a change in the pattern of local or systemic spread in vaccinated or immune animals, and, if so, which individual branches of the immune system are responsible for these changes? (2) Does immunity act simultaneously or equally on the global bacterial population, and are there intra- or inter-organ differences and heterogeneous traits in the control of individual bacterial subpopulations? (3) At which stages of the infection do different components of the immune system affect different parameters of infection dynamics? Answering these questions will illustrate how the humoral and cellular branches of immune responses control the dynamics of infection at a multiscale level, from direct interaction of bacteria with cells to the broader patterns of spread and distribution within organs and the whole body. This will provide unprecedented insight into how a highly protective experimental S. enterica vaccine works in vivo.
Acknowledgements and funding
Work on S. enterica in the Mastroeni and Grant laboratories is funded by the Biotechnology and Biological Sciences Research Council (BBSRC), including grant BBS/B/02266; the Wellcome Trust, including grant 081743/Z/06/Z; and the Medical Research Council, including grant G0801161. We thank the anonymous referees for their advice. We apologise to the authors of the many interesting and important papers that we were not able to cite in this review.






