Pathophysiology of proteinuria
Nephrology Dialysis Transplantation-Educational for kidney and blood pressure related disorders http://www.ndt-educational.org/
Proteinuria is defined as the abnormal excretion of serum proteins in urine, principally albumin. The recent 2102 KDIGO Guidelines for chronic kidney disease (CKD) evaluation, classification and management have highlighted the importance of albuminuria as a key dimension in risk assessment (Ref. 1). In healthy subjects, <30 mg/day of albumin are excreted in urine. Persistent urinary albumin excretion between 30 and 300 mg/day reflects early kidney injury, while albuminuria above 300 mg/day is indicative of more severe kidney injury and is associated with higher risk of kidney disease progression.
The origin of proteinuria can be: (1) glomerular, resulting from alterations in the integrity of the glomerular filtration barrier; (Reference Carroll and Temte2) tubular, from diminished reabsorption of normally filtered, low molecular weight proteins; or (Reference Reiser3) overflow of large loads of filtered proteins (Ref. Reference Carroll and Temte2). In this review, we will focus on glomerular proteinuria, since this is the most common form of proteinuria. Although albumin is the most abundant urinary protein found in patients with glomerular proteinuria, other plasma proteins including transferrin, immunoglobulins and vitamin D-binding protein may also be excreted (Ref. Reference Reiser3). The glomerular filtration barrier is composed by the fenestrated endothelial cells, the glomerular basement membrane and the interdigitating foot processes of podocytes that wrap around glomerular capillaries (Fig. 1). The gaps between the foot projections of podocytes are occupied by a mesh-like structure, the slit diaphragm, formed by podocyte cell-surface proteins; blood is filtered through these structures. Podocyte foot process effacement, slit diaphragm disruption and podocyte injury or loss result in increased permeability to proteins and infiltration of normally non-filtered proteins, leading to proteinuria.

Figure 1. The glomerular filtration barrier, proteinuria and tubular injury. An injured glomerular filtration barrier, usually as a consequence of podocyte injury, results in passage of huge amounts of plasma proteins into the urinary space. Such protein overload activates tubular cells to secrete chemokines and subsequent leukocyte infiltration.
Proteinuria and renal disease
Experimental and clinical studies have shown that proteinuria may accelerate the progression of CKD (Refs Reference D'Amico and Bazzi4, Reference Iseki5), being strongly associated with renal outcome in patients with non-diabetic or diabetic nephropathies (Refs Reference Breyer6, Reference Peterson7). Moreover, proteinuria is an independent predictor of both overall and cardiovascular mortality (Ref. Reference Irie8). Several clinical trials and meta-analyses have demonstrated that conventional anti-proteinuric therapy, mainly angiotensin converting enzyme inhibition or angiotensin II receptor antagonism, results in better preservation of renal function, providing evidence for a pathological role of proteinuria in the progression to end-stage renal failure (Refs Reference Jafar9, Reference Jafar10).
A number of conditions cause glomerular proteinuria. Genetic disorders in proteins important for the maintenance of podocyte cytoskeleton structure, glomerular basement membrane integrity or glomerular endothelial function may result in proteinuria (reviewed in (Ref. Reference Menon, Chuang and He11)). Diabetic nephropathy is the most frequent cause of proteinuria, although patients with focal segmental glomerulosclerosis or membranoproliferative glomerulonephritis also present proteinuria. On the other hand, a number of diseases cause nephrotic syndrome. Minimal change nephrosis is the most common disorder found in children, while membranous glomerulonephritis is most frequently seen in elderly subjects (Ref. Reference Ruggenenti, Perna and Remuzzi12).
Proteinuria-induced kidney injury: the role of chemokines
In recent years, the pathological effects of glomerular ultrafiltrate of serum proteins have been reported. It is important to note that the selectivity of the glomerular barrier in proteinuric glomerular diseases decreases in line with renal function. Abnormal passage of serum proteins across the glomerular filtration barrier may induce podocyte injury, resulting in further increase in proteinuria (Ref. Reference Abbate13). However, most evidence highlights the key role of tubular epithelial cells in the progression of renal damage as a consequence of the proteinuric state. Overexposure to filtered proteins induces a series of injurious responses in tubular cells, including apoptosis (Ref. Reference Ohse14), complement synthesis (Ref. Reference Tang15) and expression of vasoactive and fibrogenic mediators (Ref. Reference Zoja16). Importantly, proteinuria also promotes the expression and release of chemokines by tubular cells (Ref. Reference Anders17).
Chemokines comprise a group of small soluble signalling proteins, with a molecular weight of 6–14 kDa. Chemokines promote kidney tubulointerstitial inflammation (Ref. Reference Chung and Lan18). In the kidney, chemokines are implicated in the recruitment and activation of all circulating leukocytes (Fig. 1). Data from patients with proteinuric nephropathy and experimental models of protein overload have shown that interstitial infiltrates are more abundant in tubules where protein reabsorption is more active (Ref. Reference Zoja19).
Chemokines are necessary for leukocyte trafficking from the peritubular capillary lumen into renal interstitial space. In brief, in a first step the endothelium up-regulates the expression of adhesion molecules (e.g., intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion protein 1 (VCAM-1)) and selectins. These molecules are required for the efficient rolling and adhesion of leukocytes (Ref. Reference Segerer, Nelson and Schlondorff20). The endothelium also up-regulates chemokine presentation structures (such as glucosaminoglycans), which increase the endothelial capacity to bind chemokines (Ref. Reference Weber21). In a second step, chemokines exposed by endothelial cells promote the activation of leukocyte-expressed integrins, triggering further interaction with the endothelial adhesion molecules, resulting in the arrest and firm adhesion of these inflammatory cells to the endothelial surface (Ref. Reference Langer and Chavakis22). In a subsequent step, leukocytes pass between adjacent endothelial cells and arrive to the renal interstitium (Ref. Reference Imhof and Aurrand-Lions23).
Chemokine release in response to protein overload is polarised towards the basolateral compartment of tubular epithelial cells. This directional release could explain the tubulointerstitial leukocyte infiltrate reported in the initial phases of proteinuric disorders. Once infiltrated into the interstitial space, leukocytes not only secrete additional chemokines and profibrotic factors, but also amplify the release of chemokines by tubular cells via humoral factors, including IL-1β, IL-6 and tumour necrosis factor alpha (TNF-α) and also via cell-to-cell-dependent interactions through lymphocyte function-associated antigen 1 (LFA-1), ICAM-1 and CD40L/CD40 (Refs Reference Kuroiwa24, Reference Sanz25). Thus, the release of chemokines (CCL2) in cultured tubular cells exposed to elevated concentrations of albumin was increased by co-stimulation with T lymphocytes and mononuclear cells (Ref. Reference Kuroiwa24). In the same line, a positive association between tubular CCL2 expression, macrophage infiltration, and proteinuria was observed in patients with diabetic nephropathy (Ref. Reference Mezzano26). Chemokines produced by both infiltrating leukocytes and tubular cells may promote further interstitial infiltration of T cells and monocytes. These events raise the possibility that a positive feedback loop exists between tubular and infiltrating cells, which amplifies and perpetuates the inflammatory response in advanced phases of proteinuric state (Ref. Reference Chung and Lan18).
Chemokines and chemokine receptors
Currently, there are more than 50 known chemokines and 20 chemokine receptors (Fig. 2) (Ref. Reference Zlotnik and Yoshie27). Most chemokines have four characteristic cysteine residues, and depending on the motif displayed by the first two cysteines, they can be classified into four different classes: CXC or alpha, CC or beta, C or gamma, and CX3C or delta. The only exception to the four cysteine rule is lymphotactin, which has only two cysteine residues (Ref. Reference Ansel28). A further division of CXC family can be made depending on the presence of a Glu–Leu–Arg sequence (ELR motif) preceding the first cysteines, namely ELR+ CXC and ELR − CXC chemokines, with antagonic properties on angiogenesis. The ELR+ CXC subfamily is composed by CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7 and CXCL8 (Ref. Reference Murphy29). The ELR − CXC subfamily is composed by CXCL9, CXCL10, CXCL11, CXCL12, CXCL13, CXCL14, CXCL15, CXCL16 and CXLCL17 (Ref. Reference Moreno30).
Figure 2. Diagram of the human chemokine superfamily, arranged by families based on the number of cysteines. Receptors are represented by R- and their given number. Chemokines are clustered depending on their binding receptor and are represented by L- and their given number.
In addition to their structural classification, chemokines may be organised according to their functional activity in inflammatory, homoeostatic or mixed function. Homoeostatic chemokines are involved in constitutive leukocyte activation, development, haematopoiesis, tissue repair and angiogenesis (Refs Reference Moore31, Reference Quackenbush32). Inflammatory chemokines are involved in the migration and activation of leukocytes and play a key role in several inflammatory diseases (Ref. Reference Strieter33). Most of the CCL-chemokines and all of the ELR+ CXC chemokines are included in this inflammatory group.
Chemokines mediate their effects through seven-transmembrane G-protein-coupled receptors. Chemokine receptors have been termed CXCR, CCR, CR and CX3CR based on the chemokine class they bind (Fig. 2) (Ref. Reference Zlotnik and Yoshie27). This type of receptors consists on a single polypeptide chain with an extracellular N-terminal domain and a cytoplasmatic C-terminal domain. The C-terminus and the intracellular domains cooperate to activate the G-protein. Chemokine receptor signalling is a complex process which activates different intracellular pathways (discussed below in more detail) (Ref. Reference Wong and Fish34). Many different chemokines bind to the same receptor and vice versa. Chemokines are able to associate between them, forming di-, tetra- or multimeres. Moreover, chemokines can be both pleiotropic and redundant in their effects and expression. The apparent redundancy and complexity of the chemokine/chemokine receptor system is the reason of the high effectiveness, flexibility and site-specificity of the system (Ref. Reference Mantovani35).
Role of chemokines and chemokine receptors in proteinuria
In recent years, the knowledge of the role of chemokines and their specific receptors in the course of proteinuria has improved (Refs Reference Schlondorff36, Reference Tipping and Holdsworth37, Reference Yamagishi38). Several chemokines have been found to be responsible for leukocyte infiltration in proteinuria-associated renal diseases (Table 1).
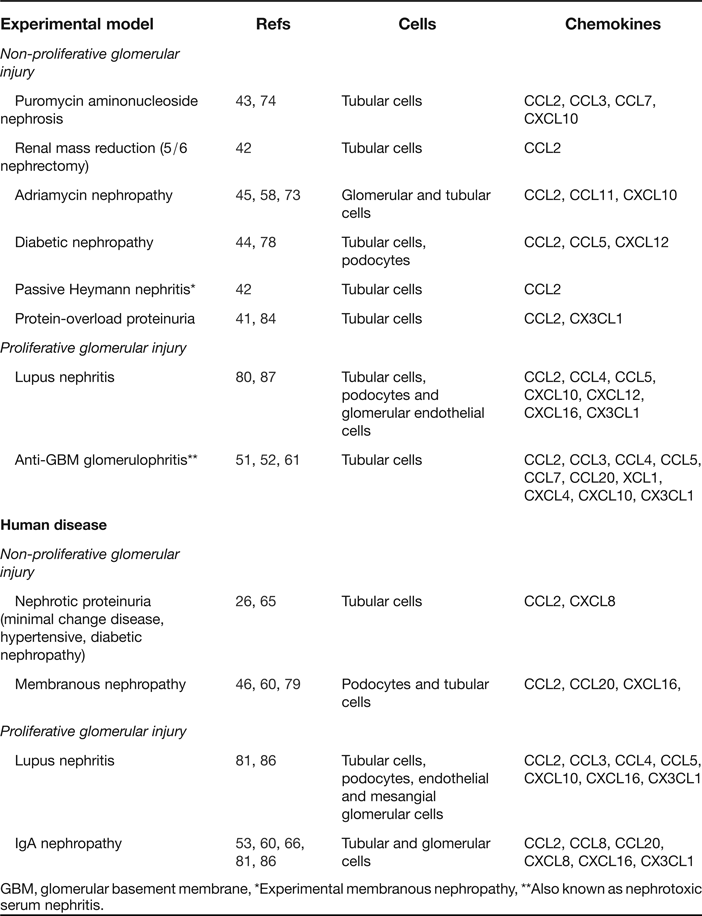
Table 1. Examples of chemokine expression in experimental and human renal diseases associated with proteinuria
GBM, glomerular basement membrane, *Experimental membranous nephropathy, **Also known as nephrotoxic serum nephritis.
CC-Chemokines
Chemokine (C–C motif) ligand 2 (CCL2), also known as monocyte chemoattractant protein 1, is one of the most widely studied chemokines. CCL2 is involved in the recruitment of monocytes and lymphocytes at sites of inflammation through the activation of several receptors, mainly CCR2 (Ref. Reference Jimenez-Sainz39). Albumin induces production of CCL2 by tubular epithelial cells (Ref. Reference Takaya40). Increased renal expression of CCL2 has been reported in experimental models of protein-overload proteinuria (Ref. Reference Eddy and Giachelli41), 5/6 nephrectomy and passive Heymann nephritis (Ref. Reference Donadelli42), puromycin aminonucleoside nephrosis (Ref. Reference Ou, Natori and Natori43), and also in patients with diabetic nephropathy and idiopathic membranous nephropathy (Ref. Reference Mezzano26). In these studies, CCL2-positive staining was principally observed in the cortical tubules and correlated with macrophage accumulation, proteinuria, and decline of renal function (Ref. Reference Chow44). In the mice model of adriamycin nephropathy, which is characterised by proteinuria and interstitial fibrosis, an increased expression of CCR2 was observed (Ref. Reference Wu45). In crescentic glomerulonephritis, the number of glomerular and interstitial infiltrating CCR2-positive cells was associated with CCL2 expression. Furthermore, these proteins were identified as biomarkers of an adverse renal function outcome in membranous nephropathy (Ref. Reference Yoshimoto46). It has been suggested that determination of chemokines in the urine may provide a more precise overview of the inflammatory status of the kidney. Thus, urinary CCL2 levels closely reflected renal inflammatory state and correlated with albuminuria and inflammatory infiltrates in children with IgA nephropathy and focal segmental glomerulosclerosis (Ref. Reference Wasilewska47). Urinary CCL2 levels were also identified as biomarkers of prognosis in CKD (Ref. 48) and lupus nephritis, and is a useful non-invasive approach in disease screening (Ref. Reference El-Shehaby49).
Chemokine (C–C motif) ligand 5 (CCL5), also known as RANTES, and its specific receptors CCR1, CCR3 and CCR5 have also been associated with proteinuria-related renal injury. Albumin and other filtered urinary proteins, such as IgA and IgG, can induce up-regulation of CCL5. As compared with CCL2, CCL5 induction is delayed but sustained in time, remaining increased for days. CCL5 was expressed by tubular epithelial cells during proteinuria, and its expression was associated with interstitial CCR5-positive mononuclear cells (predominantly CD3-positive T lymphocytes) and fibrosis in patients with acute interstitial nephritis (Ref. Reference Segerer50). Increased CCL5 levels have been found in kidneys of autoimmune nephritis, which is characterised by proteinuria and monocyte recruitment (Ref. Reference Xie51).
After induction of nephrotoxic nephritis, proteinuric mice exhibit increased glomerular mRNA expression of CCR1, CCR2 and CCR5 along with tubular mRNA expression of CCL2, CCL5 and CXCL10 (Ref. Reference Schadde52). CCR1 expression as well as CCR5+ inflammatory cells has been reported in renal biopsies from patients with IgA nephropathy (Ref. Reference Wagrowska-Danilewicz, Danilewicz and Stasikowska53). CCR1 was also up-regulated in the kidney cortex of mice with immune complex glomerulonephritis (Ref. Reference Anders54). In diabetic nephropathy, hyperglycaemia increases reactive oxygen species (ROS) production, which, in turn, up-regulates CCL2 and CCL5 expression by tubular cells, resulting in amplification of the harmful effects associated with proteinuria (Ref. Reference Elmarakby and Sullivan55). Interestingly, after initial remission, urinary CCL5 levels were identified as predictors of renal failure in patients with glomerulonephritis (Ref. Reference Tian56).
CCL11 is an inflammatory chemokine involved in eosinophil recruitment in several inflammatory diseases (Ref. Reference Pease57). Recently, Pereira et al. found elevated levels of CCL11 in experimental adriamycin-induced focal and segmental glomerulosclerosis, which is associated with proteinuria (Ref. Reference Pereira58).
CCL20/LARC is a homoeostatic chemokine that activates the CCR6 receptor (Ref. Reference Cook59). The CCR6/CCL20 pair has been found in association with the recruitment of T and B cells in IgA nephropathy, membranous nephropathy, and crescentic glomerulonephritis (Ref. Reference Welsh-Bacic60). CCR6 was overexpressed in tubular epithelium of these inflamed kidneys. Moreover, induction of nephrotoxic nephritis resulted in CCL20 up-regulation, T cell recruitment, renal injury, albuminuria and renal function impairment (Ref. Reference Turner61). In this study, disruption of the CCR6 gene aggravated renal injury by reducing the recruitment of anti-inflammatory FoxP3 + CD4+ regulatory T cells and IL-17-producing CD4+ T cells. CCR6 and CCL20 mRNA expression was also increased in kidney biopsies of patients with a progressive course of IgA nephropathy compared with those with stable renal function, suggesting a potential role of CCR6/CCL20 system in mediating progressive renal disease (Ref. Reference Villa62). In the same paper, treatment with telmisartan, an angiotensin II receptor blocker (ARB), reduced CCR6 mRNA and protein expression in nephritic rats, which identifies CCR6 as a potential key mediator of ARB-related protective effects (Ref. Reference Villa62).
The role of Duffy antigen receptor for chemokines (DARC) in proteinuria-associated glomerular diseases is not well-known. DARC is a non-specific receptor for many chemokines, including CCL2, CCL5 and CXCL8. DARC is highly up-regulated in renal injury, mainly on peritubular capillaries (Ref. Reference Segerer63). DARC facilitates renal recruitment of macrophages, T cells and neutrophils. However, in mouse models of prolonged renal inflammation, genetic elimination of Darc enhanced development of renal infiltrates during the early phases of interstitial and glomerular diseases (Ref. Reference Vielhauer64). Therefore, new studies are necessary to understand the specific function of DARC in vivo.
CXC-chemokines
CXCL8, also known IL-8, is an inflammatory chemokine that activates CXCR1 and CXCR2 and promotes neutrophil infiltration and activation in proteinuria-associated renal diseases. CXCL8 expression was increased in tubules from patients with heavy proteinuria and non-proliferative glomerulopathy (Ref. Reference Tang65) and IgA nephropathy (Ref. Reference Yokoyama66). In vitro studies in tubular epithelial cells have shown that albumin increases CXCL8 through the activation of the nuclear factor-kappaB (NF-κB) transcription factor. CXCL8 urine levels have been used as a marker of renal disease progression (Refs Reference Ni67, Reference Khajehdehi68). Thus, increased urinary levels of CXCL8 were observed in IgA nephropathy, lupus nephritis and membranoproliferative glomerulonephritis (Refs Reference Yokoyama66, Reference Wada69). In lupus nephritis, urinary levels of CXCL8 were used as a marker of disease activity (Ref. Reference Rovin70). Urinary CXCL8 levels were higher in patients with glomerular leukocyte infiltration than in those without infiltration (Ref. Reference Wada69). Moreover, patients in remission showed lower CXCL8 levels than patients with idiopathic nephrotic syndrome in relapse, and levels correlated with proteinuria values (Ref. Reference Souto71).
CXCL10, also known as interferon-inducible protein (IP-10), promotes activation and recruitment of T cells, eosinophils and monocytes (Ref. Reference Lee, Lee and Song72). CXCL10 is expressed by glomerular and tubulointerstitial cells in experimental adriamycin nephrosis and puromycin aminonucleoside nephropathy and is implicated in the pathogenesis of glomerulonephritis including lupus nephritis and IgA nephropathy (Refs Reference Gomez-Chiarri73, Reference Han74). Expression levels of CXCL10 and its cognate receptor, CXCR3, were higher in urinary cells from patients with lupus nephritis as compared with controls (Ref. Reference Avihingsanon75). However, a recent study reported a decreased glomerular and tubulointerstitial CXCR3 expression in patients with lupus nephritis (Ref. Reference Lu76). In this study, glomerular CXCR3 expression was inversely correlated with the degree of proteinuria.
CXCL12 is a pleiotropic chemokine with both homoeostatic and inflammatory properties. The principal CXCL12 receptor, CXCR4, was up-regulated in kidneys from experimental glomerulonephritis (Ref. Reference Wang77). In these models, the activation of the CXCL12/CXCR4 axis promoted the migration of B cells. In a mouse model of diabetic nephropathy, an increased production of CXCL12 by podocytes was observed, contributing to proteinuria and glomerulosclerosis (Ref. Reference Sayyed78).
CXCL16 chemokine can function as a membrane-bound adhesion molecule and a soluble chemokine. CXCL16 plays a pivotal role in the development of renal injury and proteinuria through regulation of macrophage and T cell infiltration. CXCL16 expression was increased in glomeruli of patients with membranous nephropathy (Ref. Reference Gutwein79) and in tubular and glomerular cells of lupic mice (Ref. Reference Teramoto80) and in human chronic glomerulonephritis (Ref. Reference Izquierdo81).
CX3C-chemokine
CX3CL1/fractalkine is both a membrane anchored chemokine and a soluble protein that can be shed from the membrane after proteolytic cleavage (Ref. Reference Bazan82). Soluble CX3CL1 causes migration of cytotoxic T lymphocytes and macrophages, whereas the membrane-bound form promotes adhesion and enhances the subsequent migration of these cells at tubular sites (Refs Reference Chakravorty83, Reference Donadelli84). Protein overload-induced proteinuria was associated with CX3CL1 gene expression in renal tubular cells in mice (Ref. Reference Donadelli84). In cultured renal cells, albumin induces CX3CL1 gene expression through NF-κB and p38 mitogen-activated protein kinase (p38-MAPK)-dependent pathways (Ref. Reference Donadelli84). CX3CL1 expression was also increased in tubules from patients with progressive renal disease (Ref. Reference Segerer85) and in the renal interstitium of patients with crescentic glomerulonephritis, correlating with the number of infiltrating leukocytes (Ref. Reference Furuichi86). In this regard, CX3CL1 expression was associated with monocyte accumulation and severity of glomerular lesions in experimental lupus nephritis (Ref. Reference Nakatani87). CX3CR1, the CX3CL1 exclusive receptor, is expressed in several renal cells. Thus, increased CX3CR1 expression was found in the glomerular endothelium of rats with anti-glomerular basement membrane glomerulonephritis (Ref. Reference Feng88) and proximal tubules in kidneys of patients with allograft rejection (Ref. Reference Cockwell89). In renal samples from patients with glomerular renal diseases, CX3CR1 was also found to be elevated in interstitial infiltrating T cells and macrophages, both in glomeruli and interstitium, correlating with renal function decline and proteinuria (Ref. Reference Chakravorty83).
Regulation of chemokine expression in proteinuria
Under proteinuric conditions, circulating plasma proteins reach the tubular lumen. Once in the tubular lumen, filtered proteins are almost completely reabsorbed on the brush border region of the proximal tubular epithelial cells mainly via the megalin–cubilin receptor system (Refs Reference Birn90, Reference Cui91). The megalin–cubilin system binds a number of proteins including albumin, transferrin, immunoglobulins, insulin, prolactin, haemoglobin, hormones and enzymes (Ref. Reference Birn and Christensen92). Subsequent receptor-mediated endocytosis promotes the activation of several transcription factors and intracellular signalling pathways, leading to chemokine gene expression (Fig. 3). In vitro studies on proximal tubular cells have identified the specific molecular mechanisms underlying chemokine up-regulation (Ref. Reference Baggiolini93). Exposure to albumin or other plasma proteins induces a complex cascade of steps that requires the phosphorylation of MAPK pathways, including extracellular signal-regulated kinase 1 and 2 (ERK1/ERK2) and p38-MAPK, leading to the degradation of transcriptional inhibitors and the activation of transcription factors such as CAAT enhancer binding protein (C/EBP), NF-κB and activator protein-1 (AP-1) (Refs Reference Chung and Lan18, Reference Suzuki94).
Figure 3. Molecular pathways related to protein overload and chemokine expression in tubular epithelial cells.
Nuclear translocation of activated NF-κB depends on the phosphorylation and further degradation of the inhibitory subunit IκB (Ref. Reference Sanz25). Albumin activates ERK1/ERK2 and p38-MAPK, leading to IκB degradation and further NF-κB activation in the cultured tubular cells (Refs Reference Chung and Lan18, Reference Takaya40, Reference Wang95). NF-κB activation leads to the gene expression of a large array of chemokines, including CCL2, CCL5 and CXCL8 as reported both in vitro and in vivo (Ref. Reference Tang65). Thus, NF-κB activation and increased chemokine expression have been reported in several experimental models of protein overload-induced proteinuria (Ref. Reference Eddy and Giachelli41), in human diabetic nephropathy and idiopathic membranous nephropathy (Ref. Reference Mezzano26). The key role of NF-κB in proteinuria-associated renal injury has been demonstrated by using the specific NF-κB chemical inhibitors (Ref. Reference Rangan96) or gene transfer of truncated IκBα in experimental models of protein overload (Ref. Reference Takase97). These NF-κB inhibitory strategies decreased chemokine release, tubular cell damage, interstitial leukocyte infiltration and proteinuria (Refs Reference Rangan96, Reference Takase97).
Other studies revealed ROS as second messengers in protein overload-induced NF-κB activation (Ref. Reference Morigi98). Exposure to excess proteins in proximal tubular cells induced hydrogen peroxide synthesis, an effect that, together with NF-κB activation and CCL2 and CXCL8 expression, was inhibited by antioxidants and specific inhibitors of protein kinase C (PKC). This evidence suggests that protein overload-induced ROS production, nuclear translocation of NF-κB, and consequent chemokine gene up-regulation occurs downstream of PKC activation (Ref. Reference Morigi98). Albumin-mediated ROS production is also involved in the activation of Janus Kinase (JAK)/signal transducer and activator of transcription (STAT) pathway in cultured proximal tubular cells, in particular the JAK2/STAT1,5 axis (Ref. Reference Nakajima99).
In addition to the megalin/cubilin system, albumin may interact with signalling receptors, including the epidermal growth factor receptor (EGFR). It has been suggested that EGFR plays a central role in albumin derived-signalling pathways that link albumin to the activation of ERK1/ERK2 and increased expression of CXCL8 (Ref. Reference Reich100). Therefore, EGFR may amplify the response of the proximal tubular cells to albumin, acting as a positive feedback loop.
Chemokines released by proximal tubular epithelium upon exposure to urinary proteins may stimulate these cells in an autocrine fashion (Ref. Reference Izquierdo81). Secreted chemokines may interact with the specific chemokine receptors expressed in these cells, which in turn induce the secretion of additional chemokines, inflammatory cytokines and adhesion molecules. Chemokine receptor signalling is dependent on neighbouring G-proteins. G-proteins are a composed of three distinct subunits (Gα, Gβ and Gγ). Chemokine binding to its specific transmembrane receptor results in receptor aggregation, leading to the exchange of GDP for GTP on the Gα subunit and its dissociation from the Gβ–Gγ heterodimer. Chemokine receptor dimerisation or oligomerisation is a prerequisite for receptor activation and subsequent signal transduction (Ref. Reference Milligan101). The GTP–Gα complex triggers intracellular signalling either directly by regulating tyrosine phosphorylation signalling cascades, such as JAK/STAT, or indirectly by acting on effector molecules such as adenylyl cyclase or phospholipase C, leading to alterations in cAMP levels and calcium mobilisation, respectively (Ref. Reference Wong and Fish34). Chemokine receptor homo- or heterodimerisation increases this complexity, generating a complicated network of different and related signalling cascades (Ref. Reference Mellado102). Finally, this mechanism reaches another level of complexity because of the interactions between chemokine receptors and different receptors on the cell surface, in a process named transactivation, which regulates many intracellular pathways, including ERK, JAK/STAT and Pi3 K (Ref. Reference Rajagopal103).
Targeting chemokines in proteinuria
The identification of the specific chemokines, chemokine receptors, and intracellular signalling pathways related to proteinuria has provided novel therapeutic targets to attenuate renal injury. Multiple approaches have been developed to inhibit chemokine functions, including neutralising antibodies, antagonists and molecular genetic techniques (Table 2). Knowledge about chemokine structure has also allowed for the development of novel synthetic or biological analogues to inhibit chemokine-associated effects.
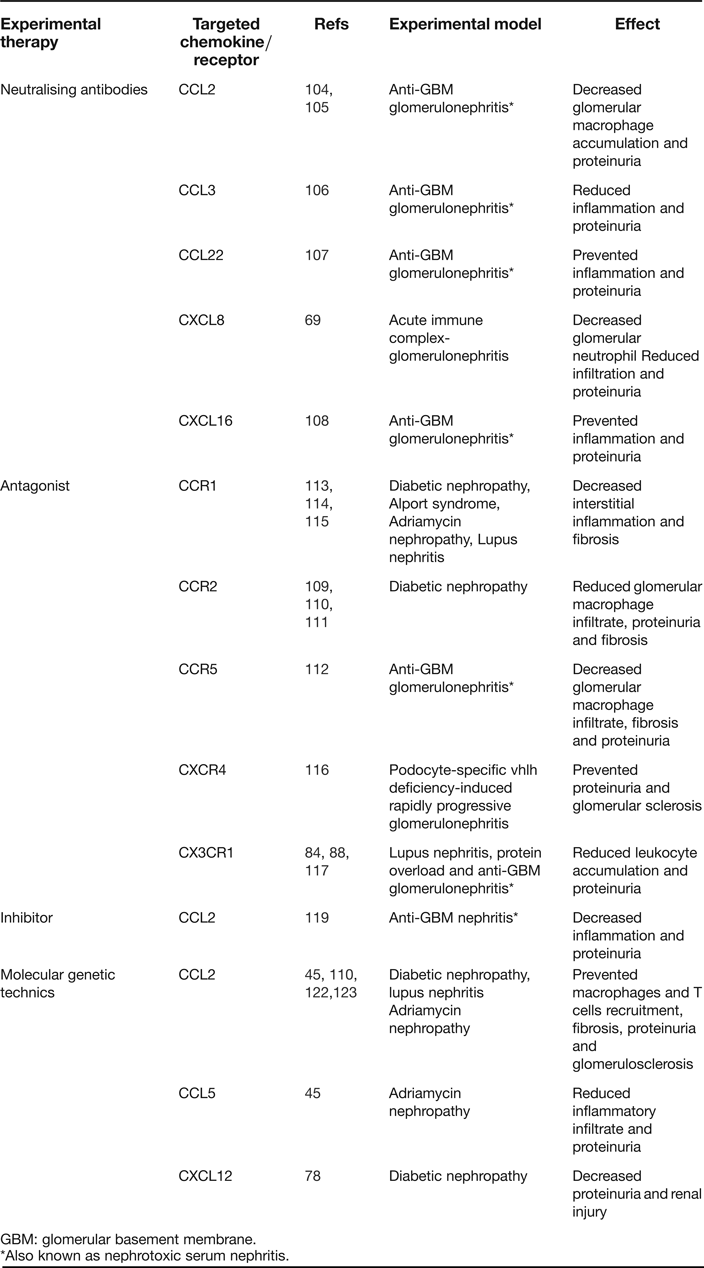
Table 2. Examples of chemokine targeting in experimental proteinuric nephropathies
GBM: glomerular basement membrane.
*Also known as nephrotoxic serum nephritis.
Neutralising antibodies
Several anti-chemokine antibodies have been used in experimental models of proteinuria. Injection of anti-CCL2-antibodies decreased glomerular macrophage accumulation and proteinuria in experimental immune glomerulonephritis (Refs Reference Wenzel104, Reference Fujinaka105). Likewise, treatment with an anti-CXCL8 antibody reduced glomerular neutrophil infiltration, prevented foot fusion processes, and normalised proteinuria (Ref. Reference Wada69), whereas CCL3, CCL22 and CXCL16 neutralising antibodies decreased inflammation and proteinuria and prevented the loss of renal function (Refs Reference Wu106, Reference Garcia107, Reference Garcia108).
Antagonists
The blockade of chemokine receptors has been used to decrease renal damage in experimental proteinuric nephropathies. In animals with diabetic nephropathy, several CCR2 antagonists (propagermanium, RS504393 and RO5234444), reduced glomerular macrophage infiltration, glomerulosclerosis, fibrosis, proinflammatory cytokine synthesis and proteinuria (Refs Reference Lee109, Reference Kanamori110, Reference Kang111). A CCR5 receptor antagonist (AOP-CCL5) reduced monocyte influx and proteinuria in experimental glomerulonephritis (Ref. Reference Panzer112). The inhibition of CCR1 receptor with the antagonist BX471 improved proteinuria-associated abnormalities in experimental models of diabetic nephropathy, glomerulosclerosis and Alport syndrome (Refs Reference Chow113, Reference Vielhauer114, Reference Ninichuk115). Furthermore, administration of a blocking antibody to CXCR4 improved the course of rapidly progressive glomerulonephritis in mice (Ref. Reference Ding116). Finally, a CX3CR1 antagonist reduced macrophage accumulation and attenuated renal injury in rodent models of lupus nephritis (Ref. Reference Inoue117), protein overload (Ref. Reference Donadelli84) and glomerulonephritis (Ref. Reference Feng88).
Inhibitors
Novel agents have been developed to inhibit chemokine or chemokine receptors. Heteroaroylphenylureas inhibit CCL2-induced chemotaxis of monocytes/macrophages both in vitro and in vivo, but do not antagonise CCL2-CCR2 interaction (Ref. Reference Laborde118). CCL2 expression is controlled to a significant extent by the transcription factor NF-κB, and therefore, treatment of rats with the NF-κB inhibitor pyrrolidine dithiocarbamate led to a decrease in CCL2 expression, reduced proteinuria, and preserved renal function (Ref. Reference Sakurai119). Currently available drugs are being studied for non-specific interference with CX3CL1. Bacoflen, a gamma-aminobutyric acid-B (GABA-B) receptor agonist, decreased inflammation in a mouse model of contact hypersensitivity. This effect was ascribed to the desensitisation of several chemokine receptors including CX3CR1 (Ref. Reference Duthey120). Rosiglitazone is also able to suppress CX3CL1 signalling through peroxisome proliferator-activated receptor (PPAR)-gamma activation (Ref. Reference Wan and Evans121).
Molecular genetic techniques
In recent years, a number of molecular genetic approaches have been developed to neutralise the inflammatory effects of chemokines. Blocking of CCL2 action with a plasmid containing a mutant of CCL2 (7ND) reduced glomerular macrophage recruitment and glomerulosclerosis in diabetic mice (Ref. Reference Kanamori110). DNA vaccination with naked DNA encoding for CCL2 or CCL5, which resulted in the induction of autoantibodies, reduced inflammatory infiltration and decreased proteinuria in the adriamycin model of nephropathy (Ref. Reference Wu45). Subcutaneous infusion of cells transfected with a vector expressing a truncated inactive form of CCL2 suppressed the development of renal inflammation in a mouse model of lupus nephritis (Ref. Reference Hasegawa122). A novel strategy that consists in the use of L-enantiomeric RNA oligonucleotides (Spiegelmer) for blocking CCL2 and CXCL12 prevented the onset of albuminuria in lupus nephritis (Ref. Reference Kulkarni123) and type 2 diabetes (Ref. Reference Sayyed78), respectively.
Multiple targeting
The chemokine/chemokine receptor network is redundant in function since many chemokines have been shown to signal through multiple receptors and vice versa. Owing to this redundancy, it has been suggested that blocking multiple chemokines or chemokine receptors may be a more effective strategy for complete inhibition of inflammatory events. In this sense, a multiple receptor antagonist (Met-CCL5) allowed simultaneous blockade of CCR1 and CCR5 in autoimmune diseases (Refs Reference Plater-Zyberk124, Reference Matsui125). Dual antagonists that target both CCR2 and CCR5 have been also developed (Refs Reference Kothandan, Gadhe and Cho126, Reference Zheng127). On the other hand, blockade of CCR2 and CXCR3 receptors was obtained by using antibodies blocking simultaneously CCL2/CCL8 (Ref. Reference Reid128) and CXCL9/CXCL10 (Ref. Reference Fagete129), respectively. More recently, a soluble CCR5-fusion protein selectively bound and neutralised all three CCR5 ligands (CCL3, CCL4 and CCL5) in vivo (Ref. Reference Sapir130). In the last year, a novel monoclonal antibody was also developed to inhibit CCL3, CCL4 and CCL5 signalling on both CCR1 and CCR5 in vitro and in vivo (Ref. Reference Scalley-Kim131). Although the neutralisation of multiple chemokine activities with these targeting strategies has provided significant therapeutic benefit in experimental autoimmune and inflammatory disorders, only one study has been reported in proteinuric conditions. Thus, combination of Spiegelmer specific for CXCL12 and CCL2 prevented proteinuria in mice with diabetic nephropathy (Ref. Reference Darisipudi132). More research is required to explore their therapeutic possibilities and effects in proteinuric conditions.
Clinical implications/applications
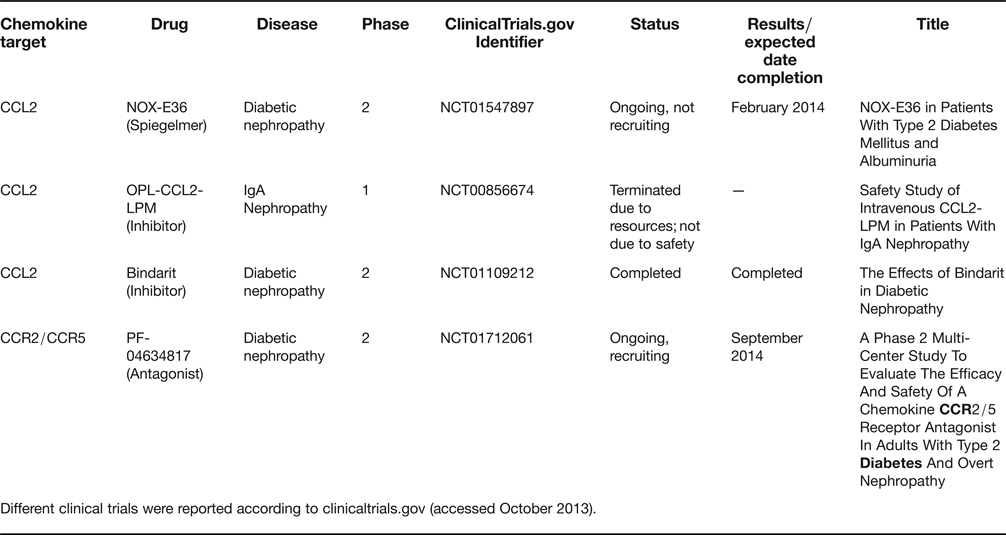
Despite the promising results of anti-chemokine therapies in animal models, the effectiveness of these approaches has only been addressed in a limited amount of clinical trials in patients with proteinuric renal diseases. The current status of clinical trials exploring anti-chemokine strategies is summarised in Table 3. To date, only CCL2 and its receptor CCR2 have been targeted in the context of proteinuric kidney disease. The therapeutic benefit of inhibiting chemokine system has only been demonstrated in subjects with lupus and active nephritis, where the CCL2 inhibitor bindarit significantly reduced albuminuria and urinary CCL2 levels in a small, proof-of-concept trial (Ref. Reference Ble133). No results have been published from a completed clinical trial testing the efficacy of bindarit in diabetic nephropathy (ClinicalTrials.gov Identifier: NCT01109212). A phase I trial of CCL2-LPM (CCL2-leukocyte population modulator) in patients with IgA nephropathy was terminated for lack of resources (ClinicalTrials.gov Identifier: NCT00856674). The CCL2 Spiegelmer NOX-E36 is undergoing phase 2 clinical trials in diabetic nephropathy to test efficacy in decreasing the urinary albumin:creatinine ratio. Enrolment has been closed and the study is expected to be completed by February 2014 (NCT01547897). A new study is recruiting diabetic patients with overt nephropathy to evaluate the efficacy of the CCR2/5 antagonist PF-04634817 to decrease the urinary albumin:creatinine ratio (ClinicalTrials.gov Identifier: NCT01712061). A further phase 2 clinical trial targeted CCL8 actions with reparixin, an inhibitor of CXCL8 receptor CXCR1 and CXCR2 activation, to prevent delayed graft dysfunction, a non-proteinuric nephropathy (ClinicalTrials.gov Identifier: NCT00248040). The trial was completed but results have not been published yet. Thus, new clinical trials are clearly needed to confirm the potentially beneficial effects of chemokine-targeted therapy in patients with proteinuria.
Table 3. Clinical trials of drugs targeting chemokines and their receptors in proteinuric kidney disease
Different clinical trials were reported according to clinicaltrials.gov (accessed October 2013).
Research in progress and outstanding research questions
Specific chemokine-targeted therapy has evolved as a novel therapeutic approach in proteinuria. Several strategies, including specific antibodies against either chemokine or their receptors have successfully decreased leukocyte chemotaxis and proteinuria in experimental animal models. However, although several anti-chemokine agents are currently being tested in clinical trials, at present there is no sufficient evidence supporting the efficacy of these therapeutic tools in patients with proteinuric diseases. Moreover, clinical trials with anti-chemokine agents should be primarily conducted in those patients where conventional therapies failed.
A number of compounds and therapeutic strategies have effectively inhibited pro-inflammatory chemokine actions in different experimental pathological conditions. However, it is important to note that not all available anti-chemokine therapies have been applied in the context of proteinuria. Therefore, we need to gain further insight into this issue. Furthermore, we need to learn more about dosing and safety of the anti-chemokine compounds and to investigate its beneficial effects singularly or in combination with conventional anti-inflammatory or anti-proteinuric treatments used in proteinuria.
Finally, despite significant advances, more biological data are needed on the mechanisms underlying chemokine regulation in proteinuric disorders. In some experimental studies, individual anti-chemokine strategies failed to demonstrate efficacy in the treatment of proteinuria (Ref. Reference Anders17). Several explanations for this limited success have been proposed, including increased complexity and functional redundancy in the chemokine signalling network. In this sense, strategies allowing for simultaneous blockade of multiple chemokine or chemokine receptors may be more effective. However, the current therapeutic arsenal to block multiple chemokine targets is limited and, to date, it has not been applied for decreasing proteinuria.
In conclusion, there is experimental evidence that chemokines contribute to the progression of renal injury induced by proteinuria. Increased protein overload activates tubular epithelium to produce inflammatory chemokines and profibrotic mediators and, despite years of intense research, much remains to be learned about this pathological process, as well as their clinical implications and potential therapeutic. However, despite a promising proof-of-concept study, the safety and efficacy of such approaches in humans remains to be demonstrated.
Acknowledgements and funding
FIS (CP10/00479, Programa Miguel Servet) and PI13/00802 to J.A.M. Fundación Conchita Rábago to A.R.N. Ministry of Science (SAF2012/38830) and Sociedad Española de Nefrologia to C.G.G. Sociedad Española de Nefrologia, ISCIII PS09/00447, REDinREN/RD12/0021/0001, Comunidad de Madrid/FRACM/S-BIO0283/2006, Programa Intensificación Actividad Investigadora (ISCIII/) to A.O., and ISCIII-Redes RECAVA (RD06/0014/0035), European Network (HEALTH F2-2008-200647), Euro Salud EUS2005-03565, cvREMOD, Fundacion Lilly, FRIAT and ISCIII fund PI10/00072 to J.E.