Background
Glioblastoma (GBM) is an aggressive brain tumour which arises from glial cells in the central nervous system (CNS) (Ref. Reference Gladson1), however, there is still very little known with regards to its cause and associated risks. Unlike other cancer types, there are only two known risk factors, one being ionising radiation as part of treatment for various conditions and the other family history. Controversy remains with respect to its precise cell of origin (Ref. Reference Gimple2). It occurs in two to three people per 100 000 per year in the adult population (Ref. Reference Rock3). The median age at diagnosis is 64 years and the disease is slightly more common in males compared with females (Ref. Reference Tamimi, Juweid and De Vleeschouwer4). GBMs can arise as primary or secondary tumours. Primary tumours are more common, accounting for 90% of cases (Ref. Reference McGuire5) and develop rapidly de novo, whereas secondary tumours occur when low-grade gliomas become more aggressive (Ref. Reference Ohgaki and Kleihues6). GBMs are highly aggressive because of their heterogeneous nature and also late detection of these tumours.
Patients present with symptoms and signs related to raised intracranial pressure and the location of the tumour. These include headache, nausea, vomiting, tiredness, seizures, limb weakness, sensory disturbance, incoordination, personality change, psychosis and altered affect (Ref. Reference Golla H7). Radiological imaging modalities such as computed tomography and magnetic resonance imaging (MRI) head scans help identify and locate the tumour. Apart from their origin the risk factors predisposing patients to developing GBM are still poorly defined. It is known that hereditary syndromes, such as tuberous sclerosis and neurofibromatosis, and exposure to ionising radiation increase the risk of developing the disease, but studies investigating the effect of environmental factors have been inconclusive (Ref. Reference Nelson8).
Brain tumours are traditionally classified according to their location and histopathological characteristics. Histopathological features of GBMs include necrosis, microvascular perfusion and rapid infiltrating growth, which often extend into the contralateral hemisphere of the brain (Ref. Reference Nørøxe9). Since 2016, the World Health Organization classification of CNS tumours also includes molecular parameters in addition to histology. The genetic status of isocitrate dehydrogenase (IDH) (Weller et al.) divided GBM into three groups: GBM IDH wild-type (IDHwt) which accounts for 90% of cases, IDH-mutant and NOS, that is, not otherwise specified, where full IDH evaluation cannot be performed. In some cases, 1p/19q and other genetic parameters were prioritised over a histological phenotype to distinguish GBM from anaplastic astrocytoma (Ref. Reference Louis10). The presence of molecular markers such as methyl-guanine methyl transferase (MGMT) can be identified in GBM tissue samples. MGMT is an important DNA repair enzyme encoded by the MGMT gene. Techniques such as methylation-specific polymerase chain reaction can identify the degree of MGMT methylation. GBMs with increased methylation express MGMT less, hence lack the DNA repair functionality which this enzyme affords (Ref. Reference Stupp11). The loss of expression of MGMT is a significant, independent prognosticator of response to chemotherapeutic agents such as temozolomide (TMZ) (Ref. Reference Hegi12). Freely available online resources such as TCGA (Ref. Reference Institute13) and cBioPortal (Refs Reference Cerami14, Reference Gao15) highlight the most common gene mutations found in GBM, with IDH, TP53 gene (TP53), ATRX (ATRX Chromatin Remodeler), TTN and PTEN (Phosphatase and Tensin Homologue)featuring strongly in this list.
For newly diagnosed patients, current standard of care involves maximal safe surgical resection followed by concurrent radiotherapy and chemotherapy with TMZ and subsequent adjuvant TMZ chemotherapy (Ref. Reference Stupp16). At recurrence, with no established standard of care (depending on individual patients and their disease history) treatment options include further surgical resection, radiotherapy and systemic therapy such as lomustine or bevacizumab, combined approaches or supportive care alone (Ref. Reference Tan17). Advanced stereotactic radiosurgery technologies such as the Gamma-Knife (Ref. Reference Crowley18), linear accelerator-based X-Knife and CyberKnife are typically used as a salvage treatment in patients with recurrent GBM to avoid further surgical procedures (Ref. Reference Yaprak19) or as a complementary approach to conventional fractionated radiotherapy (Ref. Reference Lipani20). Implementation of biomedical optics as intraoperative guidance tools in GBM has seen great success (Ref. Reference Stummer21). A recent review summarised three major optical technologies that are available clinically: fluorescence, reflectance and Raman (Ref. Reference Valdés22). Fluorescence technologies reply on fluorophores either endogenous or exogenous, whereas the latter two reply on intrinsic optical signals – reflectance technologies interrogate tissue based on light–tissue interactions and chromophores such as oxy- and deoxyhaemoglobin, and Raman detects the vibrational energies of molecular bonds in tissues. However, unlike other types of cancer where significant improvements in survival have been made, little progress has been made towards improving this in GBM over the last four decades (Ref. Reference Damiani23). This is also because GBM as a disease entity is notoriously difficult to treat using conventional pharmacological therapy. The tumour is frequently resistant to commonly used chemotherapy whereas in addition the surrounding brain tissue is susceptible to damage from adjuvant radiotherapy. Finally, the blood–brain barrier (BBB) makes delivery of drugs to the tumour site challenging (Ref. Reference Lawson24). The median survival of patients diagnosed with GBM is short – usually only about 12–18 months (Ref. Reference Gladson1). Age and Karnofsky Performance Score at diagnosis are the main prognostic factors for survival (Ref. Reference Lamborn25). The lack of effective treatment options destines GBM to be a disease of unmet medical need (Refs Reference Girvan26–Reference Wick and Kessler28).
Perhaps, the greatest challenge in developing more effective treatments for GBM is the failure of current in vitro and in vivo preclinical models to fully recapitulate GBM pathophysiology, making it difficult to predict which lead compounds will translate from the lab into successful drugs to use in the clinic (Ref. Reference Le Rhun29). Indeed, several attempts to integrate molecular-targeted agents such as Rindopepimut targeting the EGFR deletion mutation EGFRvIII (Ref. Reference Weller30), mTOR inhibitors temsirolimus (Ref. Reference Chang31) and everolimus (Ref. Reference Ma32), CDK4/6 inhibitor palbociclib (Ref. Reference Taylor33), vascular endothelial growth factor (VEGF) inhibitor cediranib (Ref. Reference Batchelor34), tyrosine kinase inhibitor galunisertib (Ref. Reference Brandes35), checkpoint inhibitor nivolumab (Ref. Reference Reardon36), etc., into GBM treatment have shown potential in conventional preclinical models but failed to pass clinical trials. Were new laboratory models developed that could mimic the in vivo response of GBM to drugs more effectively, they could substantially improve our ability to predict the safety and efficacy of substances in preclinical testing, which is essential to make the discovery of GBM therapies a more efficient process.
Here, we will discuss advantages and limitations of such preclinical models, with a focus primarily on novel in vitro/ex vivo technologies including three-dimensional (3D) (multi-) cell culture, organoid-based culture, and the application of bioprinting and microfluidics to achieve more complex tumour microenvironment mimicking plus high-throughput screening and analysis.
Preclinical models
Cell lines
For over 50 years, human tumour-derived cell (lines have been indispensable tools for basic and translational oncology in GBM. The U-251 MG and U-87 MG cell lines are among the most commonly used, both of which were generated over 50 years ago (Ref. Reference Pontén and Macintyre37). The European Collection of Authenticated Cell Cultures (ECACC) listed U-251 MG (formerly distributed as U-373 MG (ECACC catalogue number 89081403)) as pleomorphic/astrocytoid cells and U-87 MG as epithelial-like cells (Refs Reference Pontén and Macintyre37–Reference Houchens40). Tumours arising from these cell lines share some features with original tumours and established cell lines are stillbeing used for intracranial injection because of their short time to become established median survival: 30 days), which may be reflective of the initial number of cells injected (around 1 × 105 – 1 × 106 Table 1).
Table 1. Commonly used xenograft transplantation models and their associated cell lines
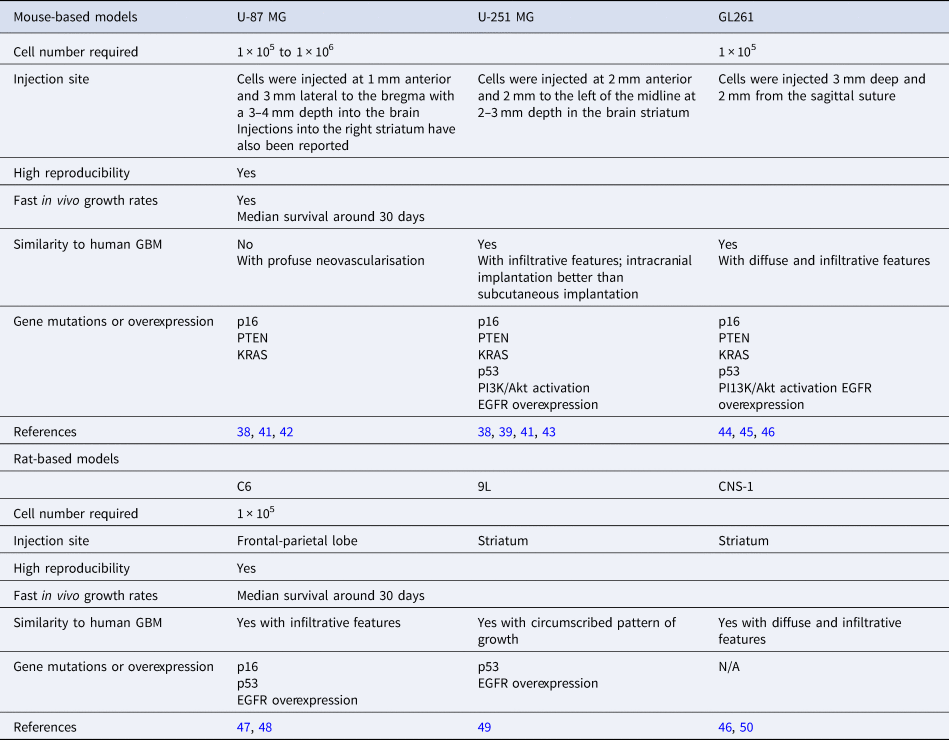
Histologically, U-251 MG induces tumours with infiltrative features such as the presence of single invading cells in the normal brain parenchyma as well as perineuronal satellitosis and glioma cells following neuronal tracks. Other GBM-associated features observed are palisading necrotic foci, microvascular proliferation, high mitotic activity and the presence of oedematous and haemorrhagic regions (Refs Reference Candolfi38, Reference Radaelli41). Tumour-bearing mice have been reported to have a median survival of 28.5 days (Ref. Reference Candolfi38). Genetic alterations such as upregulation of phosphoinositide 3-kinases (PI3Ks)/protein kinase B (Akt) pathways characteristic of GBM feature prominently in U-251 MG-induced tumours (Ref. Reference Radaelli41). However, no immunological responses to the tumour have been reported for this model. Moreover, subcutaneous and intracranial tumour models elicit different gene expression profiles (Ref. Reference Camphausen43).
In contrast, U-87 MG-induced tumours differ from human GBMs histologically. These tumours lack the characteristic diffuse, infiltrative growth pattern of GBM concomitant with a lack of necrotic foci, pseudopalisading cells and neutrophil infiltration normally associated with these tumours. Tumours appear well demarcated with a clear tumour border surrounded by reactive astrocytes (Ref. Reference Radaelli41). Mice bearing U-87 MG xenografts have reported a median survival time of 22 days (Ref. Reference Candolfi38). However, characteristic for U-87 MG is the development of tumour vasculature with homogenous and leaky vessels making them a good model for the screening of anti-angiogenic therapeutics. Genetically, U-87 MG cells have shown similarities and dissimilarities to GBM cells. The latter includes aberrant PI3K/Akt signalling and the former a wild-type tumour protein P53 (p53) background (Refs Reference Ishii51, Reference Krakstad and Chekenya52). Overall, because of its dissimilarities with GBM at the histological and genetic levels the use of U-87 MG appears to be limited to angiogenic studies of GBM. However, it has been reported that the DNA profile of the U-87 MG cell line differed from the tumour of origin (Ref. Reference Allen53); although U-87 MG still appears to originate from a GBM, it is not a true representative of its perceived tumour of origin. With several reports of cell line misidentification in the literature, this is not an uncommon issue, but one to be aware of when choosing a cell line for preclinical research (Ref. Reference Masters54). Finally, as GBM preferentially uses glycolysis for metabolism via the ‘Warburg effect’, the metabolic statuses of U251-MG, U373-MG, T98G and D54 were assessed (Ref. Reference Arthurs55). U251-MG, U373-MG and D54 mirrored mitochondrial metabolism of primary GBM cells whereas the T98G cell line recapitulated glycolysis-related metabolism of primary GBM cells. T98G was therefore recommended as the preferred model when investigating glycolysis in GBM for the identification of novel therapeutics.
Patient-derived cell lines
As well as some of the limitations associated with the cell lines described above, many were established decades ago and may have lost important features of the original tumours they were derived from. Consequently, in the last decade, scientists have taken steps to establish new patient-derived cell lines that better recapitulate the histology and genetic profiles of GBM and are used at low passage when newly isolated. A publicly available biobank of 48 new GBM cell lines, representing all four transcriptional subtypes, was developed in 2015 (Ref. Reference Xie56). There is also an increasing interest in using patient-derived glioma stem cells (glioma stem-like cells; GSCs) as they exhibit genetic and phenotypic properties which are more relevant to GBM (Ref. Reference Sampetrean and Saya57). Crucially, these GSCs showed higher resistance to conventional therapy (Refs Reference Gimple2, Reference Bao58, Reference Hubert59) and are considered as the source for not only tumour initiation but also recurrence. A range of culture systems have been established to generate GBM stem cell line from patient tumour tissue, most relying on coating of tissue culture plastics with laminin and using specialised neurobasal medium (Ref. Reference da Hora60) to maintain the stem cell-like features. A bank of 12 patient-derived, low-passage cell lines, covering the three subtypes of GBM (mesenchymal, classical and proneural) has been established recently. When grown in mice, these closely resembled the original tumours that they were generated from and have been used in various applications, for example, identification and validation of GBM- and GSC-associated therapeutics and the evaluation of novel agents for treating GBMs (Ref. Reference Stringer61) (Fig. 1). These new patient-derived cell lines add to the range and scope of available GBM models and are continuing to be an important resource for the development of clinically relevant in vitro models. On the contrary, although phenotypically and genotypically closer to their original tumour patient-derived cell lines are difficult to establish and maintain in tissue culture and tumours may take 2–11 months to grow in vivo. In addition, standardised experimental plans and procedures cannot be achieved because of the heterogeneity of tumours of the individual patients the cells derived from. Finally, cells derived from low-grade tumours generally do not grow at all in vivo.
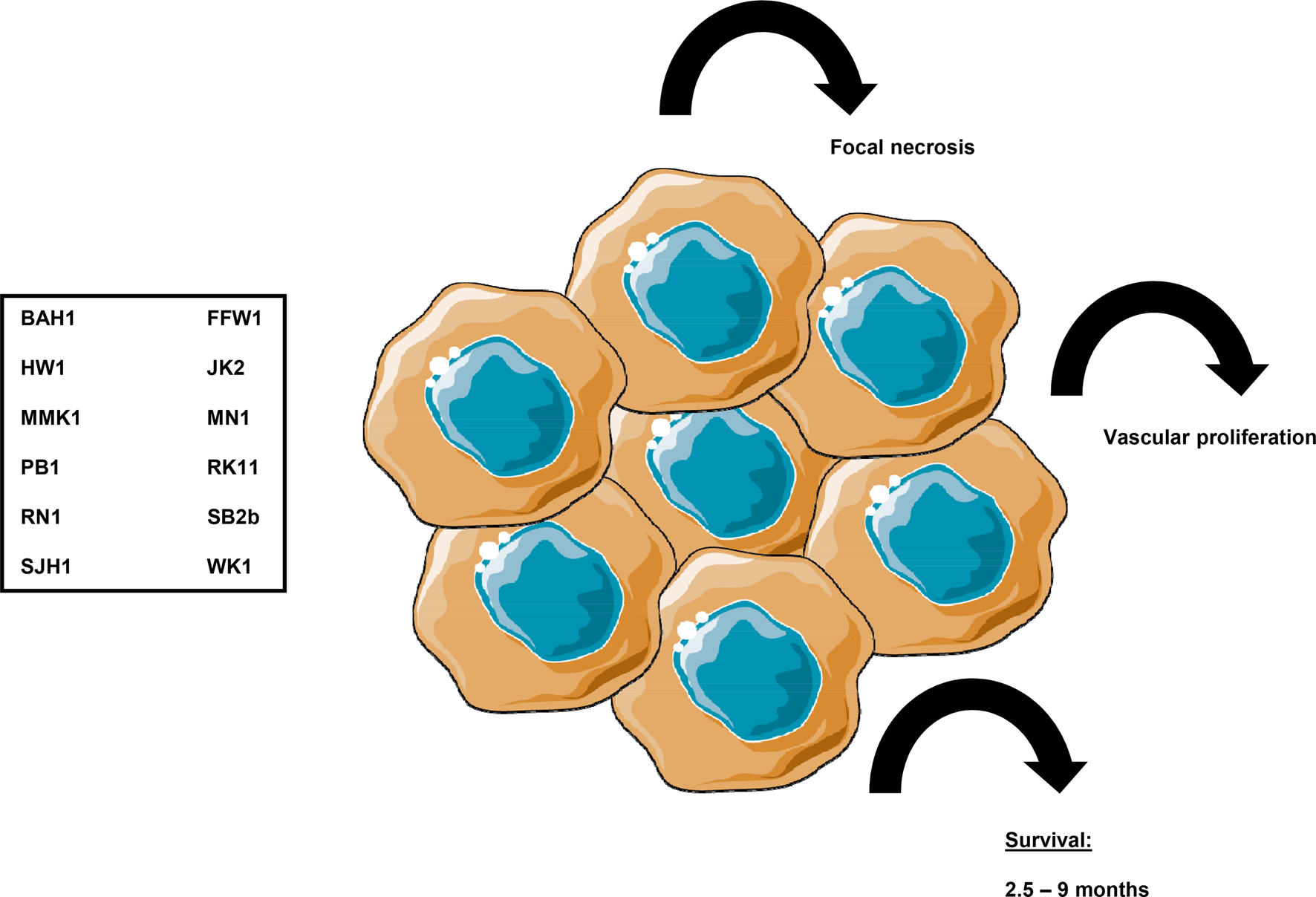
Fig. 1. Application of patient-derived cell lines for tumour formation in rodent in vivo models. Recent developments in preclinical GBM models include the use of panels of patient-derived, predefined cell lines such as the one described by Ref. (Reference Stringer61). Twelve cell lines were created shown here in the box which all were genetically diverse, yet representatives of the molecular subtypes of IDH-wild type GBMs. Xenograft tumours (represented here as accumulation of cells) developing from these cell lines were characterised by median survival times from 2.5 to 9 months, and histologically they resembled the original patient tumours they had been derived from with concomitant focal necrosis and vascular proliferation. Images were created using Smart Servier, Creative Commons Attribution 3.0 Unported License.
Xenograft transplantation models
Xenograft rodent models, predominantly mouse, have been established. Some research indicates that GBM modelling in canines is more representative than the rodent model because of the anatomical, physiological and genomic similarities to humans. Only dog GBMs exhibit endothelial proliferation, a key feature that is absent in the murine models (Ref. Reference Candolfi38). However, the high cost and ethical issues associated with using dogs mean this is rarely used. Potentially, by enhanced collaborative efforts between veterinary schools, veterinarians and GBM researchers, canine models will become more accessible for preclinical studies.
In the meantime, xenograft transplantation modelling of GBM involves subcutaneous or intracranial injection of human or mouse GBM cell lines into immunocompromised mice or rats. Tumour development and progression, drug and or radiation treatments, and overall survival can be studied to allow for cancer cell behaviour in a brain environment. Intracranial injection tends to be the preferred method as it preserves the physiological constraints of the BBB and the cerebrospinal fluid, while via subcutaneous injection, patient-derived xenograft (PDX) tumours are confined within the subcutaneous space, which is quite different from the brain microenvironment and usually fail to grow. Using this type of model, various groups have reported on the use of intravital imaging of the brain allowing imaging of GBM in situ. For this, glioma cells are labelled with green-fluorescent protein (GFP) prior to intracranial injection and an intracranial window is created to allow real-time imaging using multiphoton microscopy. This methodology has allowed investigations into the role of tumour-associated macrophages (TAMs) in tumour resistance (Ref. Reference Chen62) and vessel co-option as potential feature of resistance to anti-angiogenic treatment (Ref. Reference Seano and Jain63). Studies of cellular dynamics of migration and invasion in GBM (Ref. Reference Alieva64) have also been made possible, which is of particular interest especially in terms of determining drivers of cell dissemination and recurrence and in associated drug discovery.
The most commonly used xenograft transplantation models and their associated cell lines are illustrated in Figure 2 and their advantages and disadvantages are summarised in Table 1.
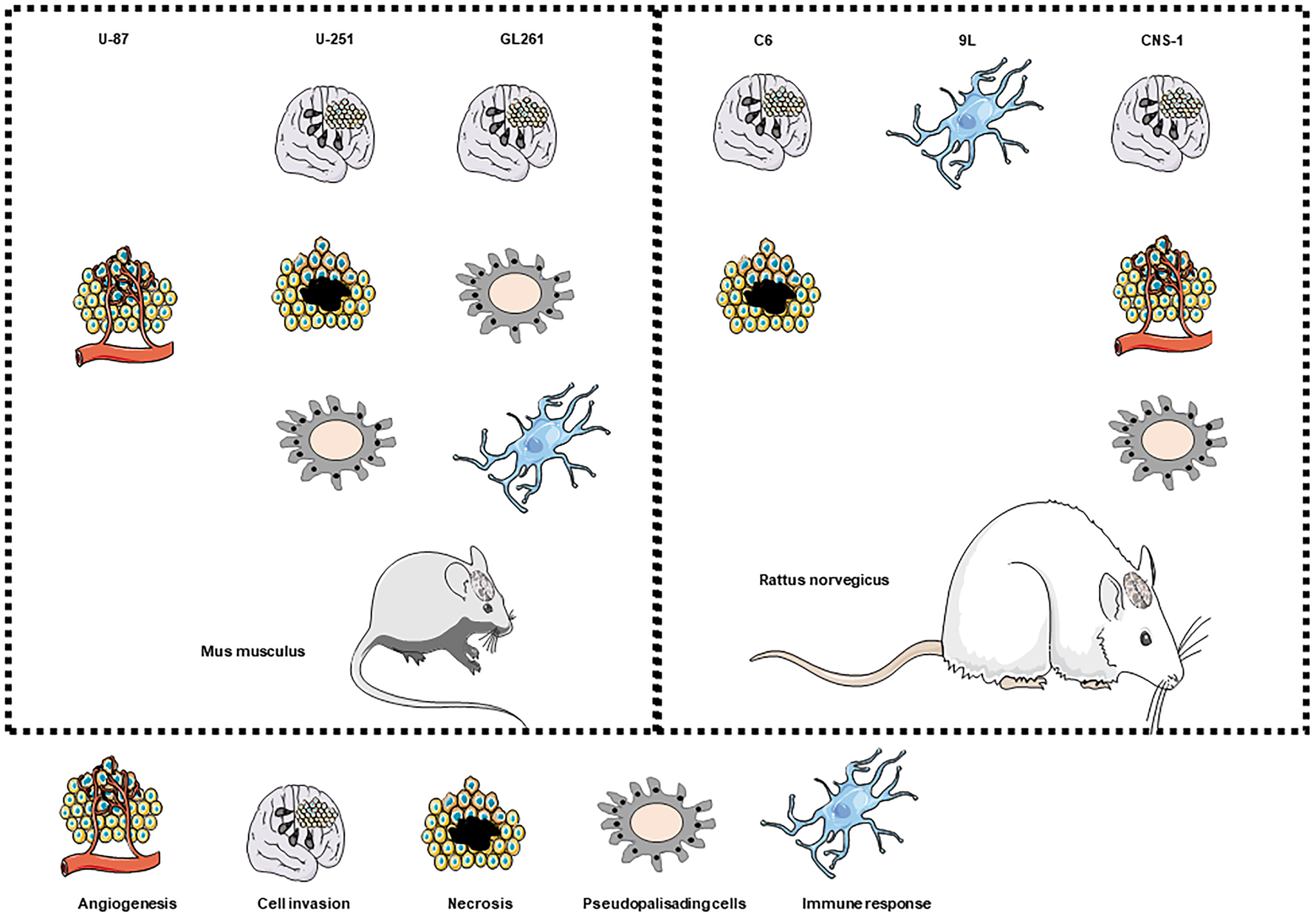
Fig. 2. Suitability of common GBM cell lines to model GBM tumour-specific characteristics when used as mouse or rat xenograft transplantation models. Cell lines used in mouse or rat (left and right panels, respectively) xenograft transplantation models are shown on the top row of each panel and their suitability to investigate angiogenesis, cell invasion, necrosis, the presence of pseudopalisading cells and immune response are shown below each cells line. Images at the bottom of the figure are representations for angiogenesis, cell invasion, necrosis, pseudopalisading cells and the immune response.
Rodent GBM models
The syngeneic mouse model GL261 is a system used in C57BL/6 mice (Refs Reference Newcomb, Zagzag and Meir44, Reference Seligman65). As such this model is not dependent on a deficient immune response and recapitulates the immunological response to GBM. Histologically, the cells induce the formation of tumours characterised by diffuse and infiltrative activity closely resembling GBM (Ref. Reference Zagzag66). In addition, perineuronal satellitosis, perivascular satellitosis, subpilar spread and cellular migration along neuronal tracks have been described (Ref. Reference Hart67). A median survival time of 31 days was noted for this cell line (Ref. Reference Candolfi38). GL261 tumours are also characterised by regions of palisading necrosis. Some key mutations such as point mutations in the K-ras oncogene and p53, as well enhanced the activation of the PI3K pathway concomitant with phosphorylation of Akt (Ref. Reference Candolfi38) have been reported. The described phenotypes especially with regards to the effect on eliciting an immune response makes this model a preferred tool when investigating immune-based therapies in GBM (Ref. Reference Van Meir68).
More recent publications highlight the advantages of a rat-based GBM model. This is especially advantageous in cases where imaging such as MRI is required to make an informed discussion of the effects of a drug in question on tumour mass and spread. Scanning mouse brains requires expensive MRI magnets to obtain anatomical features of high resolution which comes with associated financial considerations. Rat-based models circumvent these problems as rat brains are bigger therefore more easily imaged with better spatial resolution (Ref. Reference Jacobs69). The first rat model to be described in the literature was C6. Originally developed from Wistar-Furth rats this model can also be applied to breeds such as Sprague-Dawley and Long-Evans rats (Refs Reference Benda70, Reference Whittle71). Characteristic of GBM, established tumours display infiltrative invasion as well as regions of necrosis, mitotic activity and nuclear atypia (Ref. Reference Chicoine and Silbergeld72). High mutations are observed in the tumour suppressor gene p16 mirroring those observed in GBM, however, in contrast no mutations are observed in p53 (Ref. Reference Furnari73). Limitations of this model include the potential antibody response in breeds such as Wistar with loss of infiltrative behaviour and appearance of encapsulated tumours reported (Refs Reference Parsa74, Reference San-Galli75). The 9L glioma cell line was established originally in Fisher 344 rats; it has also been successfully used in allogeneic Wistar rats (Ref. Reference Kruse76). In addition to studying chemotherapeutic drugs this model is also utilised for radiation studies. CNS-1 is another commonly used rat model. CNS-1 was established in 1990 and is usually used with 1 × 105 cells, with an injection into the striatum. This model establishes tumour quickly with a median survival time of 30 days. As characteristic for GBM, the induced tumours are infiltrative and diffuse in appearance and behaviour, in addition there is perivascular spread and single cells are able to invade into normal brain parenchyma. The model is especially useful when studying the microenvironment of the tumour in relation to drug treatment (Ref. Reference Dumeule77). Tumour-associated endothelial cells have been shown to be capable of undergoing hyperplasia and necrosis characterised by pseudopalisading features making it a good model to study angiogenesis. Crucially, tumour formation also induces the infiltration of the brain parenchyma by macrophages and microglia, a prominent feature in GBM leading to tumour growth and infiltration (Ref. Reference Kielian50). As such this model is one of the models at present available to recapitulate and investigate the tumour microenvironment and the effect of chemotherapeutic drugs (Ref. Reference Dumeule77).
Zebrafish models
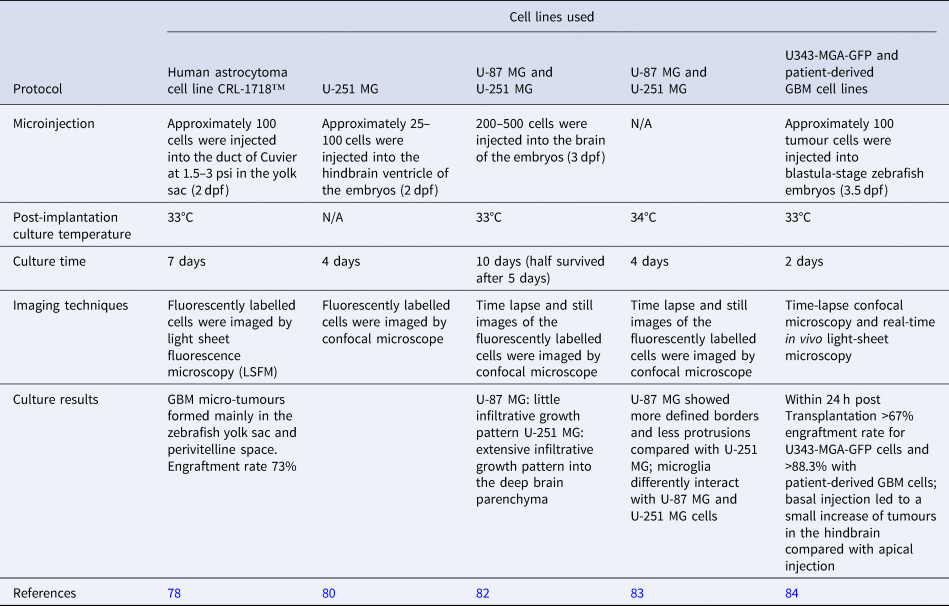
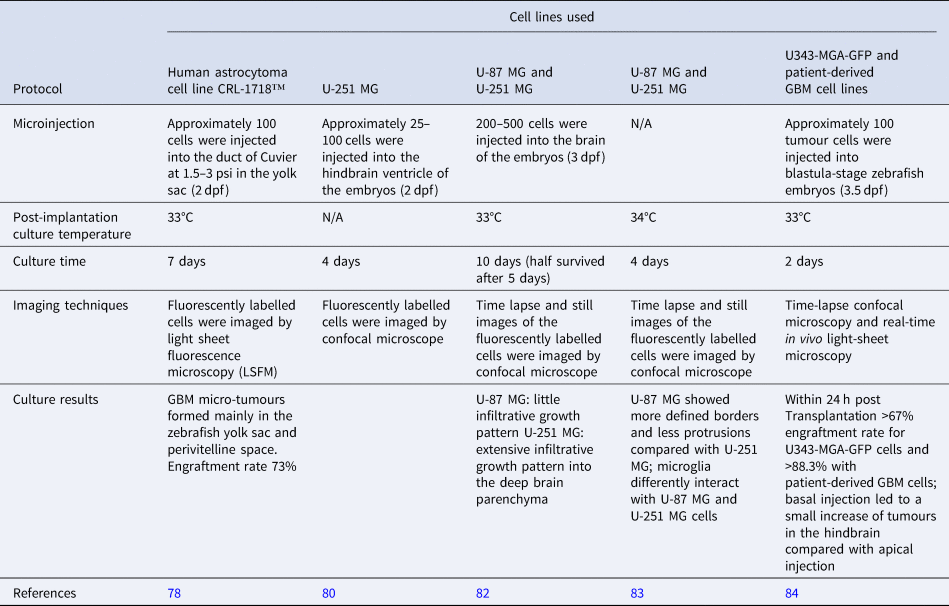
Zebrafish xenograft models are being established as an alternative model to study development and progression, cell proliferation and cellular interactions in GBM (Table 2). It has been argued that this alternative approach to studying GBM in vivo has several advantages such as the ease of generating large numbers of zebrafish offspring which can be manipulated at the embryonic stage; in addition, the fish are optically transparent during the early stages of their life cycle allowing optimum visualisation of developing tumours, and the number of animals can be up-scaled with ease for large-scale drug screens. Zebrafish can be injected with either established cell lines or patient-derived cells as xenografts. As this is usually done during the embryonic stages of the zebrafish, immunosuppression in these models is not required. Other advantages are the short time frame for tumour development once introduced, which is usually detectable within a few days rather than several weeks as in the case of the rodent models. To enhance identification of developing tumours, zebrafish larvae are microinjected with fluorescently labelled GBM cell lines and monitored by stereomicroscopy and light sheet fluorescence microscopy (Ref. Reference Vargas-Patron78). The development of microtumours can in this way closely monitor and assess activity of anti-proliferative agents. Some concerns have been raised over the fact that the zebrafish embryos are incubated at 28.5°C which may have a detrimental effect on the injected cancer cells (Ref. Reference Kimmel79). Recent studies have indicated, however, that a temperature range from 25 to 36°C xeno-injected embryo survival was up to 87.5% for the embryos incubated at the highest temperature allowing proliferation of the injected cancer cells. Other research also highlights the ability to quantify GBM proliferation, tumour dispersal, blood vessel formation and individual cell invasion (Ref. Reference Gamble80). The transparency of zebrafish embryos has been reported to facilitate imaging of tumour spread along vessels (Ref. Reference Yang81) (Fig. 3).
Table 2. Examples of zebrafish-based GBM models
dpf, days post fertilisation.
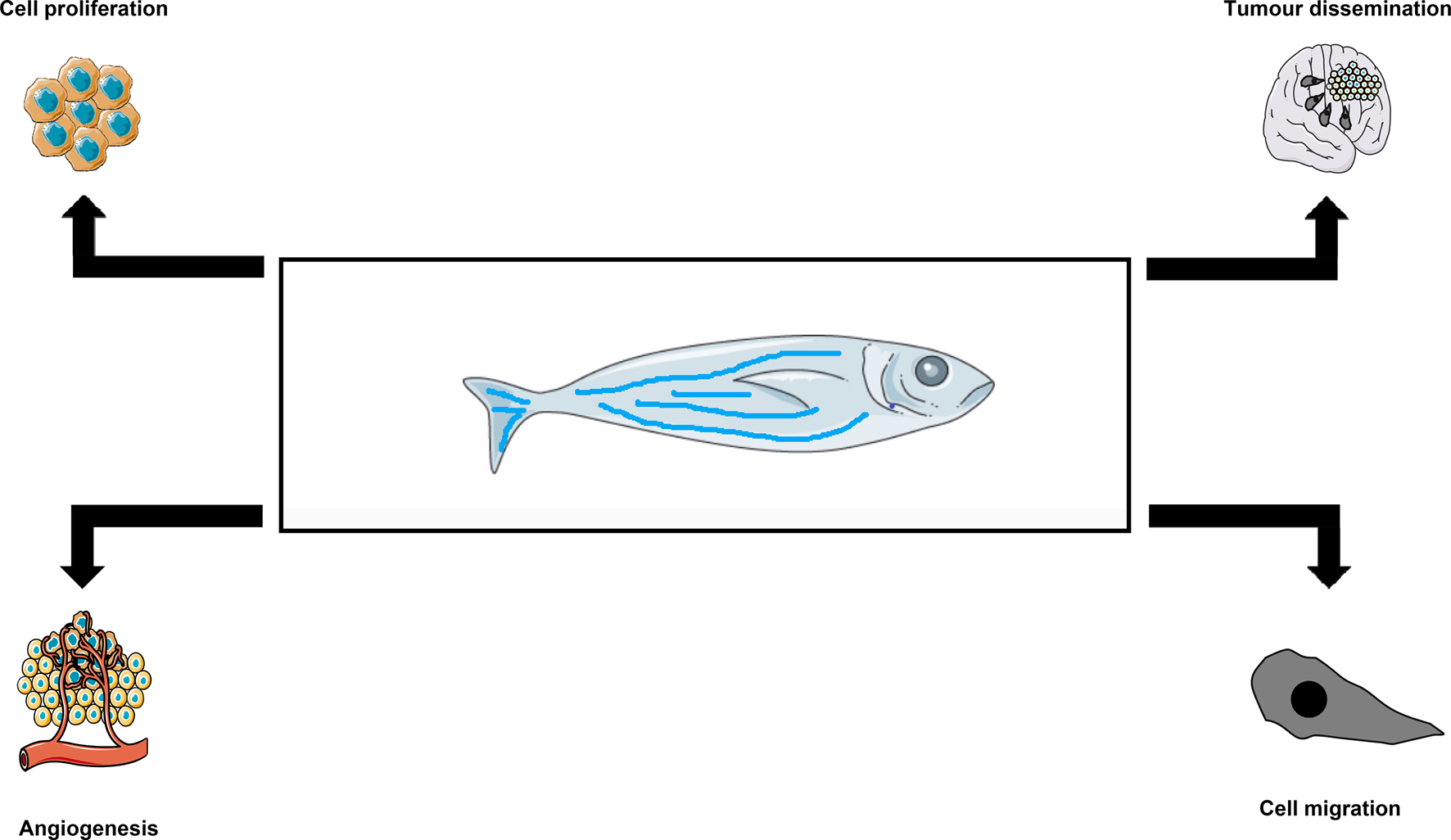
Fig. 3. Value of zebrafish models in GBM research. Zebrafish embryos are microinjected with fluorescently labelled to allow for monitoring the development of microtumours. These tumours can then be used for studies of cell proliferation, tumour dissemination, angiogenesis and cell migration.
At the cellular level, various processes can be studied, including molecular pathways, cellular processes and the role of microglia in tumour growth (Refs. Reference Hamilton83, Reference Vittori85). The most recent developments in the field highlight the possibility to use zebrafish blastomeres instead of embryos for high-throughput screening of novel therapeutic agents (Ref. Reference Pudelko84), and BBB studies for efficient characterisation of BBB penetrating anti-GBM drugs have been described (Ref. Reference Zeng82). Since this is a relatively new model to be adopted and adapted for the study of GBM preclinically and may be associated with financial implications for the set-up and maintenance of the zebrafish at various research institutions, it remains to be seen if the use of these animals is advantageous over currently used animal models. More recently, the fruit fly Drosophila melanogaster has been used as an additional experimental model for GBM (Ref. Reference Chen and Read86).
The advantages and disadvantages of the most commonly used animal models in GBM research currently, are summarised in Table 3.
Table 3. Advantages and disadvantages of commonly used animal models of GBM

Next-generation GBM modelling
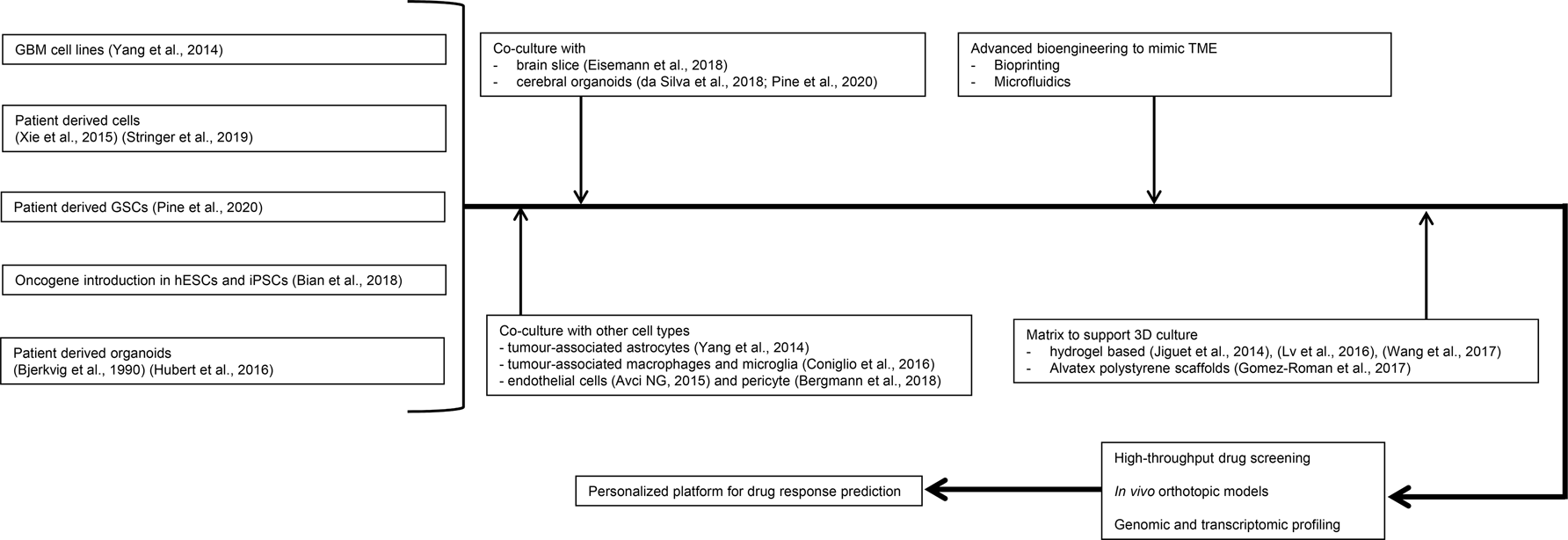
Understanding the limitations of current models, coupled to the complexity of GBM, has resulted in increased effort to develop and implement even more advanced models that better recapitulate the complex reality of GBM. Some have been reviewed specifically for radiotherapy research (Ref. Reference Caragher87) and the reader is directed to this review for additional information. Here, we discuss some successful integration of patient-derived samples and advance cell culture technologies to model GBM for drug screening.
3D cell culture
Three decades ago, multicellular organotypic spheroids were cultured successfully for the first time from human gliomas for up to 80 days. These spheroids contained preserved vessels, connective tissue and macrophages, superior to spheroids obtained from permanent cell lines (Ref. Reference Bjerkvig88). Since then, evidence of successful attempts to develop GBM-based organoid models, that is, 3D structures in which different cell types self-organise to establish appropriate cell–cell contacts and to create a microenvironment continue to exist. The chemosensitivity of GBM cells was modulated by co-culturing with astrocytes (Ref. Reference Yang81). In another co-culture system, microglia/macrophages were shown to stimulate glioma cell invasion by up to 10-fold (Ref. Reference Coniglio89). The relative expression profiles of tumour angiogenesis markers such as PECAM1/CD31 and VEGFR2 in a co-culture model of GBM cells and endothelial cells in 3D microwells were consistent with in vivo GBM studies (Ref. Reference Avci NG90). Recently, an advanced culture system using adult organotypic brain slices to study heterotypic GBM spheroids growth and invasion was developed (Ref. Reference Eisemann91). These approaches can greatly facilitate the development and/or discovery of drugs that disrupt the communication between GBM cells and others in the tumour microenvironment (TME) that enables its malignant behaviour.
Various 3D matrices have been employed to provide a microenvironment for glioma cells that was more representative of the in vivo tumour. These matrices include hyaluronic acid (HA)-rich hydrogel (Ref. Reference Jiguet Jiglaire92), collagen-based scaffolds (Ref. Reference Lv93) and poly(ethylene-glycol)-based hydrogels (Ref. Reference Wang94). These models better recapitulate in vivo conditions for drug/therapy testing compared with traditional two-dimensional (2D) culture. Gomez-Roman et al. recently developed a 3D culture system by seeding patient-derived GBM cells onto Alvatex polystyrene scaffolds. The effect of the drugs TMZ and bevacizumab on 3D cultures was similar to the effects seen in clinical trials, again suggesting that 3D cultures were more effective compared with 2D cultures at recreating the in vivo tumour response. Another advantage of this system is that it can be easily and relatively cheaply applied to high-throughput systems, including 96 well plates (Ref. Reference Gomez-Roman95).
Models employing human tissue
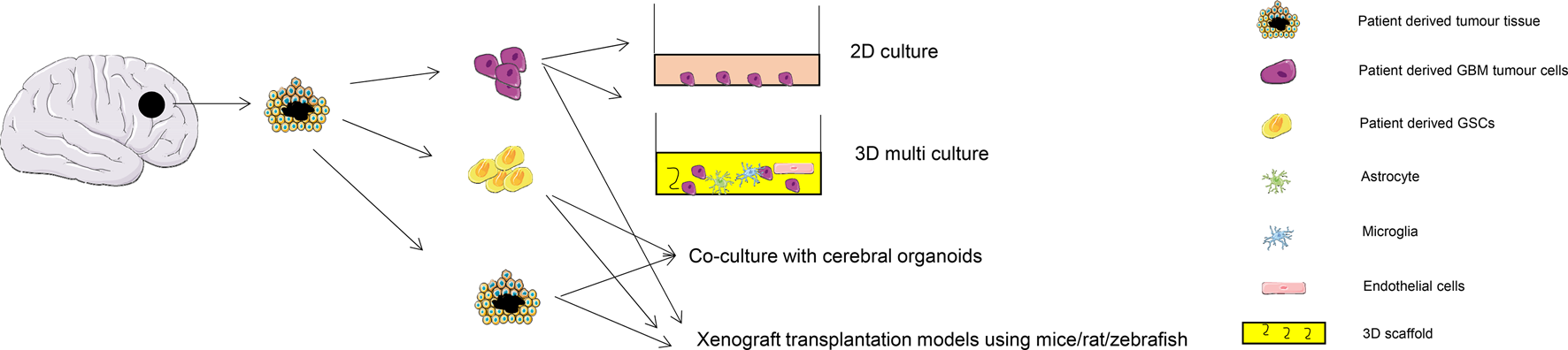
Illustrations of the types of models discussed are shown in Figure 4. PDX models of GBM are currently based on the subcutaneous or intracranial injection of either biopsied patient tumour tissue or cultured tumour spheres or stem cells into immunocompromised animals. As part of optimising this technique the growth kinetics from patient biopsies implanted via an orthotopic technique into the brains of athymic rats were analysed. Uptake of the tumour was high (96%), and the xenografts showed invasive features of the parent tumour under histological examination (Ref. Reference Wang96). There was no difference found using either fresh or cryopreserved GBM tissues in PDX engraftment suggesting a more convenient workflow for the employment of this model in preclinical GBM research as it does not have to rely on freshly obtained tumour tissue (Ref. Reference William97). For validation and proof of principle, a cohort of 40 organoid-based intracranial xenografts were compared with paired primary and recurrent gliomas. Results were encouraging as they showed the retention of intratumoral transcriptomic programmes and stem-cell-associated heterogeneity. This model was then used to test dianhydrogalactitol (VAL-083), a bifunctional alkylating agent, for treatment of GBM (Ref. Reference Golebiewska98). A library of orthotopic GBM xenograft models using surgical samples of GBM patients was reported to have successfully maintained the genomic characteristics of parental GBMs in situ. In addition, these xenografts helped to predict the pathways associated with clinical aggressiveness (Ref. Reference Eisemann91). Gao et al. developed a high-throughput screening with promising reproducibility and clinical translatability using around 1000 PDXs to predict the drug response of 62 treatments (Ref. Reference Gao99). It is hoped that these techniques could assist in making the preclinical testing of potential GBM therapy more effective in future and allow the implementation of tailored personalised therapy.

Fig. 4. Application of patient-derived materials for GBM research. Tumour tissue extracted from patient during surgery can be used as organoids (patient-derived orthotopic (PDO)) or further digested to generate tumour cells or glioma stem-like cells. PDO can be cultured using in vivo orthotopic models or ex vivo culture with cerebral organoids. Patient-derived cells can be used in conventional 2D culture, or advanced 3D culture with multiple cell types including tumour-associated astrocytes, macrophages, microglia, endothelial cells and pericytes, or as a tumour spheroid engrafted in animal models such as mice, rat and zebrafish.
Recently the clinical relevance of GSCs has been supported by increased evidence especially regarding their role in mediating therapy resistance (Ref. Reference Gimple2). A 3D culture system that supported tumour organoids derived from patient-derived primary cultures, xenografts, genetically engineered glioma models or patient samples was developed (Ref. Reference Hubert59). This model preserved both stem and non-stem GBM cell populations which had different sensitivity to radiotherapy. An elegant study by da Silva et al. created GBM organoids by co-culturing patient-derived GBM spheroids with mouse embryonic stem cell-derived cerebral organoids. The spheroids and organoids were cultured separately for 12 days prior to co-culture and upon co-culture the GBM cells infiltrated the cerebral organoids. However, although the study did investigate some of the characteristics of the resultant organoids, it did not compare them with other in vitro methods or test how the model responded to treatment with chemotherapy or radiotherapy (Ref. Reference da Silva100). Before this model could be considered for use in the preclinical testing of potential GBM therapies, both of these issues will need to be further investigated.
Interestingly, the organoid model can integrate genome-editing techniques such as CRISPR/Cas9 to introduce tumourigenic mutations. Compared with genetically engineered mouse models and PDXs using tissues, it is less expensive and time consuming to establish; while compared with 2D/3D brain cancer cell or stem cell culture, it offers the 3D organ-like structure and stromal interactions. It makes it, therefore, an attractive model for the study of the effect of tumourigenic mutations and GBM development and progression. Some successful models have been reported to recapitulate brain tumourigenesis and development: the amplification of MYC was sufficient to generate a neoplastic cerebral organoid model that could describe human CNS-primitive neuroectodermal tumour as never before in vitro nor in vivo (Ref. Reference Bian101). Simultaneously, disrupting the p53 locus and expressing the oncogenic HRAS G12V by CRISPR-mediated homologous recombination could generate more invasive tumour cells within organoids which could also be transplanted into mice and from organoid to organoid. The invasive cells were highly proliferative and expressed the stem cell marker SOX2 and GFAP at high levels (Ref. Reference Ogawa102). Further incorporation such as BBB function (Ref. Reference Bergmann103) into these organoid models can expedite their utilisation in exploring the biology and therapeutic discovery of GBM.
The use of patient samples in GBM modelling is powerful but restricted by the limited availability of starting material and the genomic instability during passaging may jeopardise their application in cancer modelling (Ref. Reference Ben-David104). Thus, it is important to assess whether these models can recapitulate patient-specific genetic and epigenetic features. Single-cell RNA sequencing can be used to characterise different GBM models and compare them with primary tumours at the cellular level. For example, the GBM cerebral organoid (GLICO) model was found to have the highest correlation with primary patient tumour compared with other three GSC-derived models: 2D glioma sphere culture, 3D tumour organoid culture and PDXs. This was evidenced by an enriched stem-like cellular state same as in primary GBM cells and the expression of NOTCH signalling. The author emphasised the importance of a neuroanatomically accurate microenvironment to GBM modelling and that this principle will likely apply for other tumours (Ref. Reference Pine105).
Additional consideration should be given to develop post-surgical residual models (Ref. Reference Rominiyi106). Samples used in mentioned pre-clinical models came from resected tumours during surgery. Given the fact that GBM is extensively heterogenic and infiltrative, it is not safe to assume the residual tumour cells after surgery can be represented by the sampled cells. How to incorporate residual cells into the pre-clinical models may hold the key to a better prediction of drug response and/or a rationale for specific targeting of post-surgical residual disease.
New technologies
High-throughput imaging and data analysis
In recent years, new technologies and associated instrumentation have been increasingly used to allow high-throughput generation and quantitative measurement of in vitro 3D GBM models. For example, cryo-imaging has been reported to enable 3D analysis of the migration and dispersal of the GFP-expressing LN-229 human glioma cell line following orthotopic injection into mouse brains. In addition to fluorescence imaging of tumour cells, algorithms were developed to aid the characterisation of blood vessels in bright-field images. Such technologies overcome the traditional in vitro confocal and multi-photon microscopy with a large volume of view, as well as in vivo methods such as MRI, positron emission tomography with high resolution and single-cell sensitivity (Ref. Reference Qutaish107). Mass spectrometry imaging was used to generate 3D dataset to map metabolites PDX models of GBM. Results revealed the increasing intensity of a series of long-chain acylcarnitines at the tumour edge corrected with a higher fatty acid metabolism which may explain the heterogeneous chemical environments within GBM tumours (Ref. Reference Randall108). An HA-based scaffold with tunable mechanical properties for culturing U118 and U-87R spheroids has been described (Ref. Reference Heffernan109). Repeated fluorescence confocal microscopy was used to track cell proliferation, dissemination and invasion in situ. Automated image analysis enabled quantitative measurement of these phenotypes through 500 μm of gel over 14 days. An ultra-high-throughput proliferation assay was tested on patient-derived GSC spheroids using commercially available culture/assay reagents. In the pilot screen, more than 3000 compounds were tested using this automation-friendly assay with high reproducibility and robustness (Ref. Reference Quereda110). There are also commercially available systems dedicated to improving high-throughput analysis system for 3D culture, one of which is the Celigo image cytometer. It has been applied for drug screening with multicellular tumour spheroid produced from U-87 MG in 384-well plates using real-time kinetic apoptosis and viability assays (Ref. Reference Kessel111). The application of the above technologies and many other emerging ones will help us to better understand the mechanism behind drug resistance of GBM and optimise the drug discovery process for patient benefits.
Bioprinting
An exciting new development is 3D bioprinting. In 3D bioprinting single cells or multiple cell types and/or biomaterials mimicking extracellular matrices are dispensed with micrometre precision to form tissue-like structures. This technology improves the simulation of the complex architecture of different tissues including GBM. Using extrusion-based bioprinting technology, a GSC culture was achieved with high-proliferation rate and stemness properties. Furthermore, the level of VEGF A secreted by the bioprinted GSCs and their in vitro vascularisation capability were higher than that of suspension-cultured cells (Ref. Reference Wang112). Unfortunately, it is a challenge to achieve high-throughput 3D culture using patient-derived tumour organoids (PTOs) as it is difficult to create large numbers of homogeneous organoids. An immersion bioprinting technology was successfully employed to overcome this by using collagen–HA bioink to minimise the bioink–well interaction. This model was used to culture two cancer cell lines HepG2 and Caco-2, as well as two GBM PTOs showing the potential using homogeneous organoids in 96-well plates that is compatible with high-throughput drug screening (Ref. Reference Maloney113).
In terms of multicellular 3D bioprinting, Yi et al. used three kinds of bioinks to achieve the co-culture of patient-derived GBM cells and human umbilical vein endothelial cells (HUVECs). This model offered a compartmentalised cancer-stroma structure, an oxygen-gradient-generating system and brain decellularised extracellular matrix (ECM), and recapitulated hallmark pathological features of human GBM such as the formation of pseudopalisades and the emergence of GSCs. The composition of the bioinks affected the sensitivity of GBM cells to concurrent chemoradiation using TMZ (Ref. Reference Yi114).
The feasibility of creating a miniaturised brain co-culturing GBM-associated macrophage (GAM) and GBM cells has been demonstrated (Ref. Reference Heinrich115). They adapted a two-step bioprinting process: first, a larger brain model encapsulating the GAMs with an empty cavity was printed, then this construct was filled with GBM cells embedded in a blend bioink consisting of gelatin methacryloyl and gelatin. Thus, the location of the tumour area was well-defined. The bioprinted cells displayed high-metabolic activity after 10 days of culture under conventional cell culture conditions. Additionally, compared with 2D culture, both GAMs and GBM cells in this model showed an upregulation of in vivo specific markers. The crosstalk between these cells was confirmed in the paracrine and juxtacrine signallings. A transcriptomic analysis of publicly available data from 159 GBM patients was performed to demonstrate the clinical relevance of gene expressions in this model. Finally, this model was used to examine the therapeutic efficacy of carmustine as well as macrophage modulating drugs AS1517499 and BLZ945.
GSCs have been incorporated with GBM cells within a unique shell/core structure. Cells encapsulated in 3% (w/v) sodium alginate were printed with a custom-made coaxial extrusion bioprinter to form shell-glioma stem cell GSC23/core-glioma cell line U118 (G/U) hydrogel microfibres. The inner diameters of which were around 400 μm with outer diameter around 850 μm. Bioprinted cells remained high cell viability after 15 days of culture. Compared with monoculture of U118 in the microfibres, the U118 co-cultured with GSC23 showed an enhanced expression of O6-methylguanine-DNA methyltransferase, more aggressive invasion phenotype and stronger resistance to TMZ (Ref. Reference Wang112).
More recently, a multi-nozzle extrusion bioprinter using RGDS-modified alginate was used to incorporate U-87 MG cells and stromal cells such as WI-38 non-immortalised fibroblasts and MM6 monocyte/macrophages. The alginate stiffness was tuned to mimic the stiffness of brain tumour tissue (1–11 kPa). A more efficient and rapid recovery of protein and RNA from cultured cells was demonstrated compared with other 3D cell culture matrices. The printed constructs also allowed fluorescent reporter analysis of protein kinase activation at the single-cell level. In addition, three different GSC lines were tested in this system, which showed over 90% viability in 7 days and maintained the expression of nestin expression even following growth factor withdrawal. Finally, drug sensitivity in the 3D-bioprinted cells were compared with those in the 2D culture. The former exhibited strong resistance to cisplatin, which agreed with its clinical performance in treating GBM. The composition of stromal cells in the multicultural system also impacted substantially the outcome of drug sensitivity (Ref. Reference Hermida116).
Above examples showed success in maintaining high cell viability and integrating suitable bioinks for high-throughput or mimicking the brain ECM, which were major concerns for bioprinting technology. The next step for GBM bioprinting would be to introduce a complex vascular system that requires higher resolution so choosing the correct bioinks and printing methods are key. Also, further transcriptional profiling of the cultured cells compared with primary tissue is necessary to match individual models to individual applications. Nevertheless, these elegant advancements are likely to contribute to the field of 3D in vitro models of GBM in future by offering relevant biomimetic characteristics and processes, promising more appropriate predictability of drug interactions.
Microfluidics
Another emerging technology is microfluidics, whereby microlitre volumes of cells and fluids may be manipulated on small, typically microscope-sized devices with etched channels. A key advantage of microfluidics is that it permits single-cell analysis as it is compatible with real-time/long-term microscopy as well as other high-resolution follow-up analyses. Through culturing U-251 MG cells on a SU-8-based microfluidic device, a robust model to mimic the GBM-associated blood vessel obstruction in vitro was achieved, which also for the first time, demonstrated the formation of a pseudopalisade-like front through three stages because of nutrient and oxygen starvation (Ref. Reference Ayuso117). Microfluidics has also been applied to isolate, enrich and characterise specific targets in GBM such as the highly mobile subpopulation from GSC-derived neurospheres (Ref. Reference Huang118), circulating brain tumour cells (Ref. Reference Sullivan119) and tumour-specific extracellular vesicles (Ref. Reference Reátegui120). Advantages include the simplified on-chip processing, sensitivity, rapid analysis time and minimal requirement of the clinical samples.
Polydimethylsiloxane (PDMS) is commonly used to fabricate chips because of its flexibility, biocompatibility, optical transparent and low cost. Integration of biomimetic hydrogels into the microfluidic chips is often used to simulate in vivo TME. For example, collagen (1.5 mg/ml) was used to encapsulate U-87 MG spheroids, which were then seeded into a 4 × 4 microfluidic array that consisted of concentration gradient generator channels to mimic drug stimulation and a precision syringe pump to generate perfusion culture. This system allowed the determination of proliferation and invasiveness of the formed spheroids under single and combined medicine. However, there are some limitations within the design: it failed to restructure the shear stress observed in vivo, and sub-channels linking the microwells to the main channel should be included to ensure the equilibrium of cytokines/chemokines in the growing cells (Ref. Reference Ma121).
Another study used matrix metalloprotease (MMP)-sensitive HA hydrogel as the backbone matrix to culture glioma cell line A-172. A polyurethane nanofibre membrane was also integrated into the device not only to support the hydrogel but also to facilitate selective diffusion of media and growth factors into the hydrogel. Diffusion of medium through the hydrogel was investigated by the fluorescence recovery after photobleaching technique. A survival rate of 80 ± 5% was measured over 7 days culture with no significant differences between static and 4-dynamic flow condition. Under static conditions, cells mostly remained a round shape and were insensitive towards remodelling of the hydrogel matrix. In contrast, cells grown under dynamic conditions developed elongated shapes, their alignment and migration phenotypes changed under VEGF stimulation (Ref. Reference Lee122).
A methacrylamide-functionalised gelatin-HA gradient hydrogel was generated via chaotic advection under a computer-controlled syringe pump on a microfluidic device with GBM PDX cells encapsulated within. This system allowed location-specific analysis of cell viability and gene expression related to poor GBM prognoses (CD44, MMP-2 and VEGFA) and endogenous HA production (HAS3). Compared with EGFR wt/PTEN − specimens, other PDX variant (EGFR +/PTEN +) showed enhanced recovery from the TKI treatment (erlotinib) only in HA-rich regions of the hydrogel. Their response to a second dose of erlotinib was also strongly influenced by local HA content. These results reflected the influence of extracellular HA in both intrinsic and acquired resistance (Ref. Reference Pedron123).
Apart from HA, another major component of the ECM in brain is chondroitin sulphate proteoglycans and their glycosaminoglycan side chains (CS-GAGs). Encapsulation of U-87 MG cells with sulphated CS-GAG hydrogels exhibited enhanced migration and cytoskeletal remodelling in a microfluidics-based migration assay, which was partially mediated by CXCL12/CXCR4 and LAR signalling (Ref. Reference Logun124).
Commercial microfluidic devices have been used in GBM research as well (Ref. Reference Park125). In addition, some attempts have been reported to apply microfluidics in the creation of high-throughput 3D models of GBM. For example, U-251 MG was tested in a pneumatic microfluidic system which allowed real-time analysis and recovery of the formed spheroids (Ref. Reference Liu126).
However, there are limitations with PDMS-based chips such as the need of expensive silicon wafer, labour intensive moulding and specific assembling by plasma bonding. Apart from the different mechanical properties of PDMS as compared with the native ECM, it was also considered to be able to absorb small molecules which will affect drug diffusion/response. Fan et al. reported a novel microfluidic device using a photo-polymerisable poly(ethylene) glycol diacrylate hydrogel for drug screening. This design comprised of 24 culture chambers and a Christmas tree-shaped channel system acted as a gradient generator. The fluorescent intensity of fluorescein isothiocyanate (MW = 150 000 Da) and 4′,6-diamidino-2-phenylindole (MW = 277 Da) were used to measure the diffusion efficiency of the platform. With an optimal seeding density of 210 cells/mm2, U-87 MG cells formed 3D spheroids in this device and remained high viability after 7 days. The synergy and antagonism between pitavastatin and irinotecan were analysed on these cells (Ref. Reference Fan127). Later the same group improved the chip design by adding diffusion gaps (600 μm wide) between each microfluidic channel to prevent cross-channel interference and demonstrated the new design's capability of culturing primary cancer cells derived from GBM patients as 3D spheroids (Ref. Reference Akay128).
Microfluidics has also been used to study the interaction between different cell types in GBM TME. Patient-derived GSCs in Matrigel and HUVECs in fibrin gel were co-cultured in a chip to mimic the vascular niche in GBM. Side-by-side validation of this microfluidic model and in vivo orthotopic mice PDX model was performed for the first time, which confirmed the physiologically relevance of this model (Ref. Reference Truong129). Another study engineered a device reconstructing the GBM tumour niche of GBM cells, TAMs and endothelial cells. Using this set-up, the role of EC–macrophage interactions was highlighted to investigate the reason for failure of current anti-angiogenic therapy in GBM (Ref. Reference Cui130).
Most current applications of this technology reach beyond the study of GBM biology. For example, Olubajo et al. (Ref. Reference Olubajo131) reported the use of microfluidics to culture human GBM tissue. A total of 128 tissue biopsies from 33 patients were maintained for an average of 3 days with only 11.3% viability lost and no significant histological differences compared with fresh counterparts. Importantly, tissues showed higher viability in this ex vivo culture were associated with poorer clinical outcomes. The microfluidic device used in this study also succeeded in maintaining many other tissues highlighting the versatility and applicability of this technology (Ref. Reference Aldape132).
Conclusion
GBM can steal decades from a patient's life. Surgery is rarely curative and better adjunct and combination treatments are needed, however, treatment options lag significantly behind those available for most other forms of cancer. This is because of the highly heterogenous and infiltrative nature of these tumours, which is reflected in poor advances in the development of chemotherapeutic drugs in comparison with other cancer types. Despite extensive investment, few chemotherapeutic breakthroughs have been made in almost half a century. In a recent paper, Cancer Research UK published seven key challenges in improving patient therapy against primary brain tumours (Ref. Reference Aldape132), in which the values of advanced pre-clinical models were highlighted. Indeed, to find the most effective way to accelerate progress in the pursuit of much needed and long overdue new therapies, research must include the move from overreliance on outdated 2D cell line models towards the more sophisticated preclinical models discussed here that have the potential to be revolutionary. Advanced 3D cell culture using patient samples combined with bioengineering technologies such as bioprinting and microfluidics offers an animal-free approach to study GBM with the possibility to focus on specific cellular composition such as GSCs and to mimic BBB and TME in the culture system, which is important to consider when predicting drug response and resistance (Fig. 5). Another major advantage of these in vitro/ex vivo models is their potential of high throughput with a fraction of cost/time compared with animal models, which can be explored further when developing personalised platforms for individual patient. GBM is known to have high inter- and intra-tumoral heterogeneity, each anatomical area of the GBM tumour has different tissue stiffness, cellular composition and TME. Thus, to achieve clinical application of these models, further characterisation and systematic evaluation of various platforms is warranted to help researchers choose the best model for their intended purpose. Key information to have before using any model would be if it can recapitulate patient-specific genetic and epigenetic features, transcriptomic programmes and intratumorally heterogeneity, and if so, how long in culture it can serve as patient avatar for preclinical precision medicine. Collaborations will be needed across the fundamental research, translational and drug discovery studies and clinical applications to increase the chances of those diagnosed with this devastating condition to optimally benefit from present and future developments.
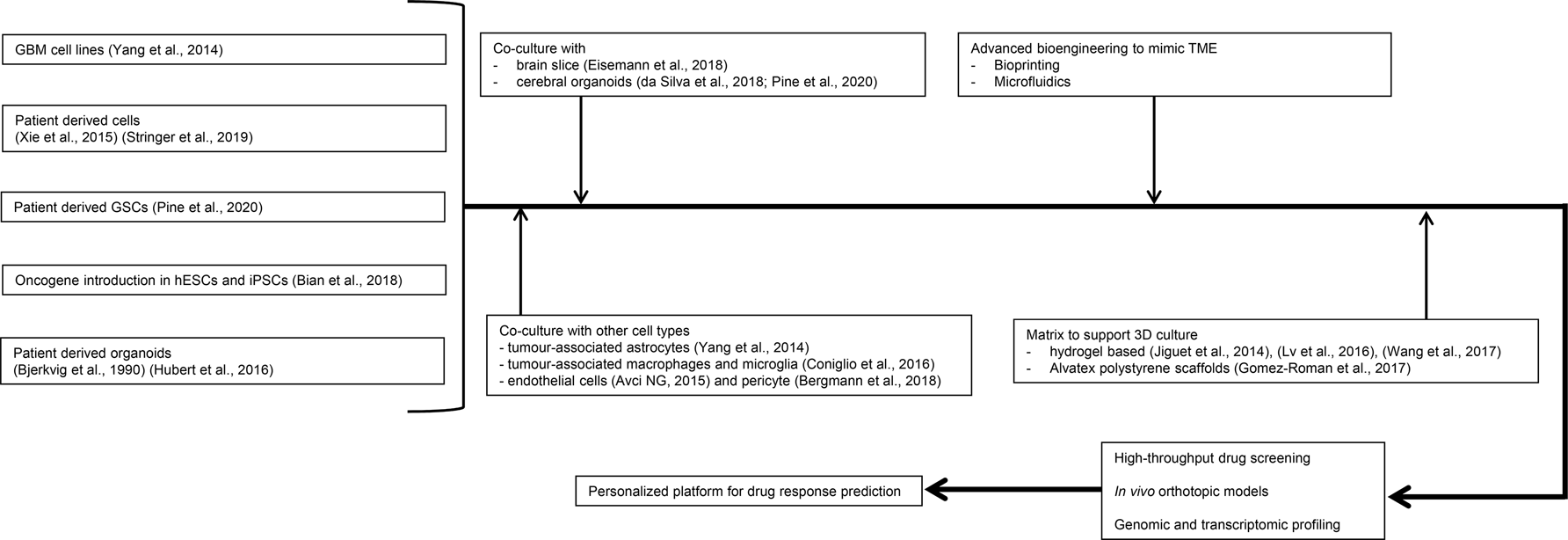
Fig. 5. Potential design of advanced glioblastoma (GBM) models for precision oncology. A shift from using conventional GBM cell lines to patient-derived materials/glioma stem-like cells has been seen. Culture system can be completed with other cell types in the TME at either the cellular level or organoid level. Controllable synthetic matrix can also be used in the culture system. Bioprinting and microfluidic technologies can be integrated to support more detailed and complex structures to mimic GBM physiology with the potential of high throughout and real-time monitoring. Samples in these advanced systems will need to be checked at the genetic and histological levels to confirm their relevance with primary patient tumours. There will then be potential for application in clinical setting in a precision medicine approach.
Acknowledgements
The authors thank members of the Speirs group for helpful comments.
Author contributions
The first draft of the manuscript was written by PL, SG, VS and ABR. DJ and PV-B contributed to subsequent sections. The manuscript was further refined by all authors. All authors read and approved the final manuscript.
Financial support
This study was supported by the University of Aberdeen Development Trust and a University of Huddersfield PhD studentship (PV-B).
Conflict of interest
The authors declare that they have no competing interests.