


Megakaryocytes (MKs), the precursors of platelets, are derived from pluripotent haematopoietic stem cells (HSCs). Every day approximately 1 × 1011 platelets are produced by the cytoplasmic fragmentation of MKs, a level of production that can increase 10- to 20-fold in times of demand and an additional 5- to 10-fold under the stimulation of thrombopoietin-mimetic drugs (Ref. Reference Lichtman1). MKs are fairly rare: within the normal human marrow, only 1 in 10 000 nucleated cells is a MK. The hallmark of the MK is its large diameter (50–100 µm) and its single, multilobulated, polyploid nucleus. The external influences that impact megakaryopoiesis include a supportive bone marrow stroma consisting of endothelial and other cells, matrix glycosaminoglycans, and a number of hormones and cytokines, such as thrombopoietin, stem cell factor and stromal-cell-derived factor-1.
In the canonical pathway of haematopoietic lineage development (Refs Reference Kanz2, Reference Nakahata, Gross and Ogawa3, Reference Akashi4, Reference Reya5, Reference Ogawa6), the HSC gives rise to two major lineages: the common lymphoid progenitor (CLP) (Ref. Reference Kondo, Weissman and Akashi7) and the common myeloid progenitor (CMP) (Ref. Reference Akashi4) (Fig. 1). The CLP then generates lymphocytes (NK, T and B cells), whereas the CMP produces the granulocyte/macrophage progenitor and the megakaryocyte/erythroid progenitor (MEP) (Refs Reference Debili8, Reference Papayannopoulou9). Other evidence suggests that the MEP can arise directly from the HSC to give rise to the erythroid and MK lineages without a CMP intermediate (Refs Reference Adolfsson10, Reference Adolfsson11). The existence of this alternate pathway is under debate. For example, Forsberg and colleagues showed that Flt3(+) early progenitors retain MK and erythroid potential in vivo at both the population and clonal levels (Ref. Reference Forsberg12). In either case, the first cells fully committed to the MK lineage are BFU-MKs (burst-forming unit megakaryocytes), which give rise to a more differentiated MK progenitor, termed colony-forming unit megakaryocyte (CFU-MK). Committed MK progenitors can be identified by a distinct surface immunophenotype as a population of CD9+CD41+FcγRloc-kit+Sca-1+IL-7Rα−Thy1.1− Lin− cells in murine bone marrow. This population is restricted to producing MKs and platelets both in vitro and in vivo (Ref. Reference Nakorn, Miyamoto and Weissman13).
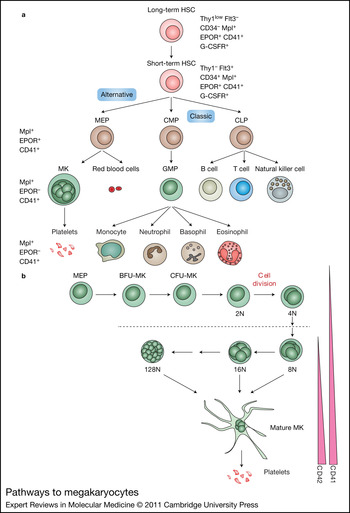
Figure 1. Pathways to megakaryocytes. (a) The HSC gives rise to all blood cell lineages. In the classical model, the first lineage commitment step of HSCs results in a separation of CMPs and CLPs. The MEP, the precursor of MKs and erythroid cells, is derived from a CMP. In the alternative model, the HSC directly gives rise to the MEP before restriction to myeloid or lymphoid lineages. (b) MK progenitors, including the MEP, BFU-MK and CFU-MK, proliferate and differentiate into platelet-producing mature MKs. During this process, MKs undergo endomitosis to increase their size and DNA content. In murine cells, the DNA content can increase up to 128N. Simultaneously, expression of the MK-specific markers CD41 and CD42 is upregulated. Abbreviations: BFU-MK, burst-forming unit megakaryocyte; CD41, glycoprotein IIb/IIIa or αIIbβ3 integrin receptor; CFU-MK, colony-forming unit megakaryocyte; CLP, common lymphoid progenitor; CMP, common myeloid progenitor; EPOR, erythropoietin receptor; Flt3, FMS-like tyrosine kinase 3; G-CSFR, G-CSF receptor; GMC, granulocyte/macrophage progenitor; HSC, haematopoietic stem cell; MEP, megakaryocyte erythroid progenitor; MK, megakaryocyte; Thy1, thymus 1 (low, low surface antigen; –, none detectable).
Megakaryopoiesis is first observed in the embryonic yolk sac where it is closely associated with primitive and definitive erythropoiesis (Refs Reference Tober14, Reference Tober, McGrath and Palis15, Reference Xie16). Primitive MEPs are seen as early as E7.25, indicating that primitive haematopoiesis is bilineage in nature. The first glycoprotein-Ib-positive cells, corresponding to MKs, can be seen in the yolk sac at E9.5, whereas platelets can be detected in the embryonic bloodstream beginning at E10.5. At this time, the number of MK progenitors begins to decline in the yolk sac and expand in the fetal liver. Fetal platelets are extremely large, with a diameter 1.6 times larger than adult platelets, and contain a large amount of RNA. This feature is also observed in patients with immune thrombocytopenic purpura and highlights the similarity between embryonic megakaryopoiesis and adult stress megakaryopoiesis.
MKs and the endomitotic process
One of the most characteristic features of MK development is endomitosis, a modified form of mitosis in which the DNA is repeatedly replicated in the absence of cytoplasmic division. During the endomitotic phase, each cycle of DNA synthesis produces an exact doubling of all the chromosomes, resulting in cells containing DNA contents ranging from 8N to 128N in a single, highly lobulated nucleus. Endomitosis is not simply the absence of mitosis, but is rather a series of recurrent cycles of aborted mitosis (Ref. Reference Vitrat17). The cell cycle kinetics of endomitotic cells is also unusual, with a shortened G1 phase, a relatively normal S phase of DNA synthesis, a short G2 phase and a very short modified mitosis phase (Refs Reference Harker18, Reference Ebbe19). During the last phase, chromosomes condense, the nuclear membrane breaks down and mitotic spindles form upon which the replicated chromosomes assemble. However, individual chromosomes fail to complete their normal migration to opposite poles of the cell, the spindle dissociates, the nuclear membrane re-forms around the entire chromosomal complement and the cell again enters the G1 phase. Endomitosis departs from a normal mitotic cell cycle at cytokinesis, when furrow invagination aborts short of cell abscission (Refs Reference Lordier20, Reference Lordier21).
Because endomitosis is a modified form of the cell cycle, one might suspect that genes involved in normal cell cycle regulation would have an important role in endomitosis. Indeed, many of the genes that control the normal cell cycle are also required for the endomitotic cycle. For example, gain of function of cyclin D1 (CCND1) and cyclin D3 (CCND3) increases the polyploid state of MKs in vivo (Refs Reference Eliades, Papadantonakis and Ravid22, Reference Sun23, Reference Zimmet24, Reference Zimmet, Toselli and Ravid25), whereas loss of function of cyclin D1 in primary murine MKs induces low polyploidy (Ref. Reference Muntean26). Similarly, loss of cyclin E (CCNE) in mice leads to defective endoreplication of MKs and trophoblast giant cells (Ref. Reference Geng27), whereas increased expression of cyclin E enhances ploidy of MKs in transgenic animals (Ref. Reference Eliades, Papadantonakis and Ravid22). In the same manner, it is likely that genes that normally restrict cell cycle progression also restrain polyploidisation of MKs. Evidence in support of this model includes observations that increased expression of p21, induced by loss of SCL/tal1, blocks endomitosis of murine primary MKs (Ref. Reference Chagraoui28) and knockdown of p19 in human CD34+ cultures results in high polyploid MKs, whereas its overexpression causes a decrease in polyploidy (Ref. Reference Gilles29).
Because polyploid cells are genetically unstable and can progress to aneuploidy and promote tumours, organisms have developed ways of minimising this process. The prevailing model has been that cells maintain a checkpoint, which monitors the number of chromosomes and results in p53-dependent cell cycle arrest of tetraploid cells. However, a number of studies suggest that normal cells are instead protected from polyploidy by activation of a stress checkpoint, which stabilises p53 and leads to cell cycle arrest (Ref. Reference Ganem, Storchova and Pellman30). The absence of data to associate polyploidisation with cellular stress in MKs might provide an explanation as to why this lineage circumvents the cell cycle arrest that is seen in normal cells.
The spindle assembly checkpoint (SAC) is an evolutionarily conserved mechanism that ensures that cells with misaligned chromosomes do not exit mitosis. The ability of the checkpoint to monitor the status of chromosome alignment is achieved by assigning checkpoint proteins to the kinetochore, a macromolecular complex that resides at centromeres of chromosomes and establishes connections with spindle microtubules. The SAC prevents cell cycle progression by negatively regulating CDC20 and inhibiting the activation of the polyubiquitylation activities of anaphase-promoting complex (APC), which degrades proteins such as securin. Failure in the function of the checkpoint results in polyploidy (Ref. Reference Ito and Matsumoto31). Genes that are required for spindle checkpoint control include the kinases BUB1 and BUBR1, the WD40 repeat containing protein BUB3, which directs the localisation of the complex to kinetochores, and the APC inhibitors MAD2/CDC20. For example, loss of function of BUB1, BUB3 and MAD2 results in increased polyploidy (Refs Reference Nakaya32, Reference Vernole33). Moreover, although homozygous loss of Bubr1 leads to embryonic lethality, Bubr1 heterozygous mutant mice displayed splenomegaly and extramedullary megakaryopoiesis with a marked increase in polyploidisation (Ref. Reference Wang34). It is important to note that haploinsufficiency of BUBR1 selectively induced polyploidy of MKs, underscoring the unique sensitivity of MKs to this process.
The chromosome passenger complex (CPC) includes Aurora-B (AURKB), inner centromere protein (INCENP), survivin (BIRC5) and borealin (CDCA8). The CPC localises at centromeres in metaphase and then translocates to the midzone late in mitosis (Ref. Reference Ruchaud, Carmena and Earnshaw35). The CPC is important for the recruitment and proper localisation of SAC proteins and is required for spindle checkpoint function. Based on this important function, one might expect that alterations in the CPC would affect MK development. Indeed, changes in survivin expression have been associated with aberrant MK polyploidisation: overexpression of survivin in normal murine bone marrow progenitors antagonised MK polyploidisation, growth and maturation (Ref. Reference Gurbuxani36), whereas loss of survivin in primary bone marrow progenitors resulted in increased polyploidy of MKs (Ref. Reference Wen37). These findings suggest that the tight regulation of survivin expression is important for MK development.
The function of Aurora-B in endomitosis has also been under intense study. Geddis and Kaushansky found that Aurora-B was expressed and properly localised during endomitosis of human MKs derived from CD34+ cells (Ref. Reference Geddis and Kaushansky38), whereas Zhang and colleagues reported that Aurora-B was absent or mislocalised in MKs derived from mouse bone marrow (Ref. Reference Zhang39). The discrepancy has been attributed to species-specific differences or to differential antibody sensitivity. Independently, Lordier and colleagues showed that inhibition of Aurora-B kinase activity induced apoptosis of low polyploid MKs and caused mis-segregation of chromosomes, mitotic failure and an increased proportion of high polyploidy human MKs (Ref. Reference Lordier21).
Precise activation of Rho-GTPase-ROCK signalling and its downstream targets, such as kinesin, myosin, tubulin and actin, are of critical importance in cytokinesis (Ref. Reference Barr and Gruneberg40). Unlike diploid MKs, RhoA and F-actin are partially concentrated at the site of furrowing in polyploid MKs and inhibition of the Rho–Rock pathway causes loss of F-actin from the midzone and increases the degree of MK polyploidisation (Ref. Reference Lordier20). A direct role for regulating RhoA-ROCK signalling has recently been demonstrated for polo-like kinase 1 (PLK1) (Refs Reference Burkard41, Reference Lowery42, Reference Petronczki43). PLK1 is important for precise localisation of ECT2 (Refs Reference Burkard41, Reference Petronczki43), a GTP exchange factor for RhoA, to the central spindle. It has been reported that PLK1 expression is absent in polyploid MKs, although expression of other cell cycle proteins is similar in both populations (Ref. Reference Yagi and Roth44). Forced expression of Plk1 during MK differentiation from murine bone marrow cells was associated with decreased polyploidisation, suggesting that PLK1 is indeed a regulator of this process in differentiating MKs (Ref. Reference Yagi and Roth44).
Transcriptional regulation of megakaryopoiesis
Lineage-specific transcription factors have essential roles in the development of MKs. Many human and murine leukaemias are associated with mutations, chromosomal translocations or viral insertions in these factors (Ref. Reference Cantor and Orkin45). This finding suggests that the maintenance of a normal transcriptional programme has an important role in normal haematopoiesis. In this section, we will highlight several key MK transcription factors, such as GATA-1, FLI1 (and other E-twenty six (ETS) factors), Stem cell leukemia (SCL), Runt-related transcription factor-1 (RUNX1) and serum response factor (SRF).
GATA-1
GATA-1 is a zinc finger transcription factor that binds the consensus motif WGATAR. GATA-1 is required for normal development of MKs and platelets. Mice that lack Gata1 expression in the MK lineage (Gata1low or Gata1knockdown mice) develop MKs, but they are abnormal in many respects (e.g. reduced polyploidisation and excessive proliferation) (Refs Reference Muntean26, Reference Shivdasani46, Reference Vyas47). Restoration of cyclin D1 expression, which is diminished in GATA-1-deficient cells, resulted in a dramatic increase in MK size and DNA content (Ref. Reference Muntean26). However, terminal differentiation was not rescued, suggesting that cyclin D1 is a mediator of polyploidisation but not of terminal differentiation. The finding also confirms the notion that polyploidisation and terminal maturation can be uncoupled. The GATA-1 protein consists of two zinc finger DNA-binding domains, the N- and C-fingers, and an N-terminal activation domain. The N-finger mediates the interaction between GATA-1 and its partner FOG-1, which is also essential for megakaryopoiesis. FOG-1-deficient mice do not generate any cells of the MK lineage (Ref. Reference Tsang48). Mice that express a Gata1 mutant that fails to interact with Fog-1 show a phenotype that resembles that of Gata1-knockdown mice, suggesting that FOG-1 has GATA-1-independent functions, probably mediated through GATA-2 (Ref. Reference Chang49). Furthermore, mutations in the N-terminal zinc finger that led to the dissociation of the GATA-1–FOG-1 interaction, or to a decreased ability of GATA-1 to bind DNA, are associated with a spectrum of benign haematologic diseases in humans, including dyserythropoietic anaemia and thrombocytopenia (Ref. Reference Crispino50), grey platelet syndrome (Ref. Reference Tubman51) and congenital erythropoietic porphyria (Ref. Reference Phillips52). NuRD interacts with the extreme N-terminus of FOG-1, and this interaction is important for GATA-1–FOG-1-mediated gene activation and repression (Ref. Reference Miccio53). Moreover, mice in which the FOG-1–NuRD interaction is disrupted produce MK progenitors that give rise to significantly fewer and less mature MKs in vitro while retaining multilineage capacity capable of generating mast cells and other myeloid lineage cells (Ref. Reference Gregory54). Further studies showed that NuRD could bind and repress expression of genes characteristic of mast cell lineage. These findings suggest that the interaction between NuRD and GATA-1–FOG-1 is required to maintain lineage fidelity in MK development. The mutations in the N-terminal domain of GATA-1 will be discussed in the acute megakaryocytic leukaemia (AMKL) section of this review.
RUNX1/AML1
The complex of RUNX1 and Core binding factor beta (CBFβ) participates in programming haematopoietic ontogeny and represents the most common mutational target in human acute leukaemia (Ref. Reference Tracey and Speck55). RUNX1 and CBFβ retain high expression during MK differentiation of MEP, but are downregulated during the early phases of erythroid differentiation (Refs Reference Kundu56, Reference Elagib57, Reference Lorsbach58). The absence of RUNX1 has profound effects on the polyploidisation and terminal maturation of MKs, leading to a significant reduction in polyploidisation and platelet formation in vivo (Refs Reference Ichikawa59, Reference Growney60, Reference Putz61). Expression of the proper amount of RUNX1 is also essential for normal platelet homeostasis, because Runx1 heterozygous animals display mild thrombocytopenia and a decrease in long-term repopulating HSCs (Ref. Reference Sun and Downing62). Mutations of RUNX1 are associated with familial platelet disorder with predisposition to acute myeloid leukaemia (AML) and sporadic cases of AML (Refs Reference Nucifora and Rowley63, Reference Song64, Reference Peterson and Zhang65).
ETS factors
Several ETS factors, including FLI1, GABPα, ETS1, ETS2, ERG and TEL1, directly contribute to murine megakaryocytic development. The importance of TEL1 was highlighted by the finding that deletion of Tel1 within erythroid and megakaryocytic cells in the GATA-1-CRE::Tel1 floxed mouse strain causes a 50% drop in platelet counts with no effect on haemoglobin levels (Ref. Reference Hock66). FLI1 is also necessary for MK development. Homozygous loss of Fli1 in mice is embryonic lethal and results in severe dysmegakaryopoiesis (Refs Reference Hart67, Reference Spyropoulos68). Accumulated evidence indicates that FLI1 is a positive regulator of megakaryopoiesis (Refs Reference Hart67, Reference Spyropoulos68) and a negative regulator of erythroid differentiation (Refs Reference Ano69, Reference Athanasiou70). Fli1 −/− mice show a marked decrease in the mature:immature MK ratio compared with wild-type littermates, consistent with a critical role in later stages of MK differentiation. Another ETS factor, GABPα, is a regulator of early MK-specific genes, including αIIb and c-mpl (Ref. Reference Pang71). The ratio of GABPα/FLI1 decreases with MK maturation, consistent with a dependence of early genes on GABPα and late MK-specific genes on FLI1 for their expression. ERG is an ETS factor encoded on human chromosome 21 (Hsa21) and expressed within MKs. ERG facilitated the expansion of MKs from wild-type, Gata1-knockdown and Gata1s-knock-in progenitors, but could not overcome the differentiation block characteristic of the Gata1-knockdown MKs (Ref. Reference Stankiewicz and Crispino72). Overexpression of ERG immortalised Gata1-knockdown and Gata1s-knock-in, but not wild type, fetal liver progenitors. Moreover, high-level overexpression of ERG resulted in myeloid leukaemia in mice, with infiltration of CD41-positive cells in the spleen (Ref. Reference Salek-Ardakani73). The latter findings highlight the important role of ERG in Down syndrome AMKL.
SCL/tal-1
It is well established that SCL is important for the development of HSCs. Mice with a conditional knockout of SCL in haematopoietic cells display perturbed megakaryopoiesis and erythropoiesis with a loss of early progenitor cells in both lineages (Ref. Reference Hall74). These mice consistently show a low platelet count and haematocrit compared with controls. The finding suggests that SCL is indispensable for the development of both megakaryocyte and erythroid lineage. SCL, along with LMO2, Ldb1 and E2A, forms a complex with GATA-1 at all activating GATA elements in MKs (Ref. Reference Tripic75). In striking contrast, at sites where GATA-1 functions as a repressor, the SCL complex is absent. Important MK targets of SCL include p21 (Ref. Reference Chagraoui28) and MEF2C (Ref. Reference Gekas76). Of note, Mef2c-deficient mice display similar MK defects as SCL-deficient animals.
Serum response factor
SRF is a MADS-box transcription factor that is important for muscle differentiation. Interests in the function of SRF in MK development was kindled when Mkl1, a cofactor of SRF, was discovered as part of the t(1;Reference Eliades, Papadantonakis and Ravid22) translocation in acute megakaryoblastic leukaemia (Ref. Reference Ma77). The importance of SRF in MK maturation and platelet formation was revealed by two groups that used Srf floxed mice and PF4-Cre mice or MX-Cre mice (Refs Reference Halene78, Reference Ragu79). Srf-deficient mice showed a reduction in platelet count and an increase of MK progenitors in the bone marrow (BM) by fluoresence activated cell sorting (FACS) analysis and CFU-MK. These mice also have defects in platelet function.
MKs and the myeloproliferative neoplasms (MPNs)
The MPNs include the ‘classical MPNs’ chronic myeloid leukaemia (CML), polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF) (Ref. Reference Dameshek80) as well as chronic eosinophilic leukaemia/hypereosinophilic syndrome, chronic myelomonocytic leukaemia, Ph-negative atypical CML, chronic neutrophilic leukaemia (CNL) and systemic mastocytosis. Many studies have demonstrated multilineage clonal haematopoiesis in the different MPNs (Refs Reference Fialkow, Gartler and Yoshida81, Reference Gilliland82, Reference Adamson83), consistent with the notion that MPNs originate in the HSC compartment.
Hyperproliferation of MKs and alterations in platelet counts are features of ET and PMF. ET is often discovered incidentally on blood counts in asymptomatic patients. The major causes of morbidity and mortality are bleeding or thrombotic complications and rarely evolution to AML. Bone marrow pathology displays increased cellularity with marked megakaryocytic hyperplasia. There are frequently giant MKs with increased polyploidy, but the overall morphology of MKs is fairly normal (Fig. 2). By contrast, individuals with PMF present with splenomegaly, dysplastic megakaryopoiesis and marked bone marrow fibrosis. The peripheral blood often shows teardrop-shaped erythrocytes and large platelets. The bone marrow shows increased deposition of reticulin fibres and numerous morphologically abnormal MKs with multilobed nuclei. PMF might progress rapidly to AML, with nearly 20% of patients transforming within 10 years (Ref. Reference Tefferi84).
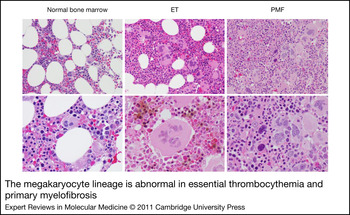
Figure 2. The megakaryocyte lineage is abnormal in essential thrombocythemia and primary myelofibrosis. Bone marrow sections from a healthy individual (left), and patients with essential thrombocythemia (ET; middle) or primary myelofibrosis (PMF; right). Top, 20 × , bottom, 40× original magnification. Note that ET MKs are typically larger than normal, increased in number and contain chromatin that resembles leaves. By contrast, PMF MKs are small, dysplastic and contain chromatin that resembles clouds. Images courtesy of Dr Sandeep Gurbuxani (University of Chicago).
Although MPNs were first described in 1951 by William Dameshek (Ref. Reference Dameshek80), the genetic basis for the disease remained elusive until 2005 when a somatic point mutation in JAK2 tyrosine kinase (JAK2V617F) was identified in 50% of ET and PMF and 95% of PV patients (Refs Reference Baxter85, Reference James86, Reference Kralovics87, Reference Levine88). JAK2 is a member of the Janus family of cytoplasmic nonreceptor tyrosine kinases, which also includes JAK1, JAK3 and TYK2. The JAK kinases have seven homologous domains (JH1–JH7) and a catalytically inactive pseudokinase domain (JH2). The predominant JAK2 mutation is a guanine-to-thymidine substitution, which results in a substitution of valine for phenylalanine at codon 617 (JAK2V617F) within the JH2 domain. The V617F mutation abrogates the autoinhibitory activity of the JH2 domain (Ref. Reference Saharinen, Takaluoma and Silvennoinen89) and results in constitutive kinase activity (Refs Reference Baxter85, Reference James86, Reference Kralovics87, Reference Levine88, Reference Ihle and Gilliland90). The mutation is present in myeloid cells, but not in germline DNA in patients with MPN (Refs Reference Kralovics87, Reference Levine88), demonstrating that JAK2V617F is a somatic mutation that is acquired in the haematopoietic compartment. When expressed in vitro, JAK2V617F, but not the wild-type protein, is constitutively phosphorylated (Ref. Reference Levine88).
How does a single disease allele contribute to the pathogenesis of PV, ET and PMF, three distinct clinical disorders? Several lines of evidence suggest that a high dose of JAK2V617F, which results in a high level of JAK2 signalling, favours an erythroid phenotype and a low dose of JAK2V617F favours a megakaryocyte phenotype. First, homozygosity for JAK2V617F is much more common in PV than in ET (Refs Reference Baxter85, Reference James86, Reference Kralovics87, Reference Levine88, Reference Scott91). Second, mutations of JAK2 exon 12, which are associated with stronger downstream JAK2 signalling compared with JAK2V617F, are found in patients with PV, but not in those with ET (Ref. Reference Scott92). Third, in vivo studies support the gene dosage hypothesis. For example, transplantation of JAK2V617F transduced mouse bone marrow cells with high-level expression resulted in a uniform PV phenotype without thrombocytosis (Ref. Reference Wernig93). Furthermore, transgenic mice that express a relatively low level of JAK2V617F, below that of wild-type allele, developed ET-like disease, whereas mice that express much higher levels caused a PV-like disease (Ref. Reference Tiedt94). A similar induction of thrombocytosis was also seen in JAK2V617F transplant recipients with the lowest level of JAK2 expression (Ref. Reference Lacout95).
Research has shown that STAT1 signalling promotes megakaryopoiesis and contributes to polyploidisation (Ref. Reference Huang96). However, a connection between STAT1 and MPN was only recently discovered. An exciting study by Anthony Green and colleagues showed that the activation status of STAT1 could predict the phenotype of JAK2V617F disease (Ref. Reference Chen97). By comparing clonally derived mutant and wild-type cells from individual patients, they showed that enhanced activation of STAT1 was associated with an ET phenotype, whereas downregulation of STAT1 activity was associated with a PV-like phenotype. It still remains to be determined whether the increased activation of STAT1 in ET is a result of change of JAK2V617F protein level.
Beyond its effects on the STAT signalling cascade, JAK2V617F was recently shown to promote changes in chromatin structure, resulting in differential gene expression (Ref. Reference Dawson98). The authors reported that JAK2 phosphorylates histone H3, leading to reduced binding of HP1α in human haematopoietic cells. Furthermore, inhibition of JAK2 activity decreased the phosphorylation of H3Y41 at certain promoters, including that of the leukaemia oncogene lmo2 (Ref. Reference Dawson98). More recently, it was found that compared with ES cells that have wild-type JAK2, levels of chromatin-bound HP1α were lower in JAK2V617F mutant cells, but these levels increased following inhibition of JAK2, coincident with a global reduction in histone H3Y41 phosphorylation (Ref. Reference Griffiths99). JAK2 inhibition also reduced levels of the pluripotency regulator Nanog, with a reduction in H3Y41 phosphorylation and a concomitant increase in HP1α levels at the Nanog promoter. These findings highlight the nuclear function of JAK2. Further genome-wide epigenetic and gene expression studies to probe JAK2 nuclear function will shed light on the pathogenesis of JAK2V617F MPNs.
This past year, four groups reported the creation of knock-in mouse models in which the mutant JAK2V617F allele was inserted into the germline in such a way that expression could be activated in a conditional fashion (Refs Reference Akada100, Reference Li101, Reference Marty102, Reference Mullally103). Three of these groups replaced the wild-type allele with the mutant murine Jak2 gene. In all these cases, animals developed similar phenotypes that mimicked PV (Ref. Reference Akada100, Reference Marty102, Reference Mullally103). By contrast, the one animal strain created with the human JAK2 gene developed an ET-like phenotype (Ref. Reference Li101). In this latter mouse, the human JAK2 cDNA was incorporated into the murine locus, an event that itself may lead to changes in gene expression and phenotype. Thus, any direct comparisons of the four lines should be done with caution.
It is interesting to focus for a moment on one of the JAK2V617F knock-in models that develops a lethal MPN (Ref. Reference Mullally103). In this model, the mice were engineered so that the wild-type exon 14 is flanked by loxP sites, with a mutated version of the exon positioned downstream in an inverted fashion. On exposure to Cre recombinase, the wild-type exon was deleted and replaced by an inverted copy of the mutated exon, leading to expression of JAK2V617F protein. By breeding floxed heterozygous mice to the E2A-Cre strain, which leads to germline excision in early embryos, JAK2V617F-expressing animals were generated. All these animals developed a lethal MPN characterised by elevated haematocrit, splenomegaly and a median survival of 146 days. Flow cytometry and histology of the bone marrow and spleen confirmed that the mice developed marked erythroid and mild megakaryocytic hyperplasia. Together, the pathological findings revealed that the mice develop an MPN that is closely reminiscent of human PV. Given that the MPN in these animals was transplantable to primary recipient mice, it was possible to identify the nature of the disease-initiating cell. JAK2 mutant stem cells exhibited a minor competitive advantage in recipients, the LSK population (and not more differentiated progenitor subsets) contained repopulating activity, and the presence of a minority of JAK2V617F LSK cells was sufficient to cause the PV phenotype. Perhaps the most fascinating aspect of the study, made possible by the ability of the LSK population to transplant disease, was the discovery that inhibition of JAK2 kinase did not eradicate the MPN-initiating population. Although treatment of primary diseased animals with the JAK inhibitor TG101348 led to significant reductions in spleen size and number of erythroid progenitors, the LSK population of TG101348-treated animals retained the ability to induce MPN in recipient mice. These studies demonstrate that JAK inhibition with this agent does not eliminate the MPN-initiating population in vivo and thus has profound implications for human therapy.
The common occurrence of JAK2 mutations in patients with MPNs underscores the importance of JAK2 signalling in the pathogenesis of these diseases. However, nearly half of ET and PMF patients do not harbour a JAK2 mutation. Shortly after the discovery of the JAK2 mutations, activating mutations in tryptophan 515 of the thrombopoietin receptor, MPL, were identified in a subset of JAK2 wild-type ET and PMF cases (Refs Reference Pikman104, Reference Chaligne105, Reference Pardanani106). These mutations occur in a subset of patients with JAK2V617F-negative ET and PMF, including 8.5% of JAK2V617F-negative ET patients (Ref. Reference Beer107) and approximately 10% of JAK2V617F-negative PMF patients (Refs Reference Pikman104, Reference Chaligne105, Reference Pardanani106). Four patients have also recently been described with somatic MPLS505N mutations (Ref. Reference Beer107), an allele that had previously been associated with inherited thrombocytosis (Ref. Reference Ding108). MPLW515-positive ET patients have higher platelet counts and lower haemoglobin levels than JAK2V617F-positive ET patients (Refs Reference Beer107, Reference Vannucchi109), and MPLW515-positive PMF patients present with more severe anaemia (Ref. Reference Guglielmelli110). Moreover, endogenous MK colonies, but not endogenous erythroid colonies, can be grown from MPLW515-positive patient cells. These findings might be because MPL is not expressed during terminal erythroid differentiation.
Because both JAK2V617F and MPLW515 activate JAK–STAT signalling, it is logical to surmise that loss of negative regulators of the pathway might also be associated with MPNs. Indeed, mutations in LNK [also known as Src homology 2 B3 (SH2B3)], a protein that downregulates JAK–STAT signalling after erythropoietin receptor (EPO-R) or MPL activation (Refs Reference Takaki111, Reference Tong and Lodish112, Reference Velazquez113), have been identified in both ET and PMF patients with a frequency between 3% and 6% (Refs Reference Lasho, Pardanani and Tefferi114, Reference Oh115, Reference Pardanani116, Reference Lasho117). LNK has a number of protein–protein interaction domains: a proline-rich amino-terminus, a pleckstrin homology domain, an Src homology 2 (SH2) domain and many potential tyrosine phosphorylation motifs (Ref. Reference Rudd118). Human LNK mutations appear to cluster in the pleckstrin homology and SH2 domains, which physically interact with the cell membrane and EPO-R/MPL/JAK2, respectively. Loss-of-function mutations of LNK were also found in JAK2 mutation negative ‘idiopathic’ erythrocytosis or PV (Ref. Reference Lasho, Pardanani and Tefferi114). Of note, mice with a complete loss of LNK display a number of features in common with human MPN, including an exaggerated response to cytokines, splenomegaly, thrombocytosis and extramedullary haematopoiesis (Refs Reference Takaki111, Reference Velazquez113, Reference Bersenev119). It should also be noted that LNK mutations and JAK2V617 are not mutually exclusive and may co-occur in the same patient (Ref. Reference Pardanani116). LNK mutations were recently shown to be acquired either early or late during the course of clonal evolution (Ref. Reference Lasho117). Other mutations identified in MPNs include TET2 (Ref. Reference Delhommeau120), ASXL1 (Ref. Reference Carbuccia121), IDH1/IDH2 (Refs Reference Green and Beer122, Reference Tefferi123), CBL (Ref. Reference Grand124), IKZF1 (Ref. Reference Jager125) and EZH2 (Ref. Reference Ernst126). The functional consequences of these mutations with respect to MPN initiation or progression are largely unknown, but are the subject of intense research.
Novel therapies for MPNs
The discovery of JAK2V617F and other JAK-STAT-activating mutations, including mutations in exon 12 of JAK2, MPL and LNK, has generated enormous interest in the development and therapeutic use of small-molecule JAK2-inhibitor-targeted therapy in these diseases. A handful of compounds are currently in clinical development for MPNs, including INCB018424, TG101348, CEP-701, SB1518 and CYT387 (Ref. Reference Tefferi and Vainchenker127). These JAK inhibitors exhibit differential inhibitory activities against the JAK family members, whereas others are less selective. For example, INCB018424 inhibits predominantly JAK1, whereas CEP-701 and TG101348 inhibit FLT3 in addition to JAK2. Data from ongoing clinical studies show that JAK inhibitors provide symptomatic relief to patients, including spleen size reduction and improvement of constitutional symptoms such as fatigue, weight loss, night sweats and fever (Refs Reference Verstovsek128, Reference Pardanani129). The improvement of constitutional symptoms by INCB018424 might be the result of a decrease in proinflammatory cytokines induced by inhibition of JAK1 (Refs Reference Verstovsek128, Reference Pardanani129). Whether this is the main mechanism of action of JAK inhibitors in general remains to be determined. Indeed, longer-term studies are required to determine the full potential of JAK2 inhibitors and to show whether they will have an impact on survival in MPNs. In addition to JAK inhibitors, several other therapies are under study for use as single agents or in combination with JAK inhibitors. These include HSP90 inhibitors (e.g. PU-H71) (Ref. Reference Marubayashi130), histone deacetylase inhibitors (e.g. pabinostat) (Ref. Reference Wang131) and PI3K–AKT pathway inhibitors (e.g. everolimus) (Ref. Reference Guglielmelli132). Given these new drugs and other lines of clinical investigation, there are many reasons to be optimistic about the prospects for improved therapies for MPNs.
Acute megakaryocytic leukaemia
AMKL is a rare subtype of AML in which the leukaemic cells are derived from the MK lineage. The three major subgroups of AMKL include infants with Down syndrome (DS-AMKL), children without DS (non-DS paediatric AMKL) and adults (adult AMKL). The incidence in the paediatric population is 1:500 children with DS and 5–7% of paediatric AML cases overall in children without DS, whereas the incidence in adults is estimated to be approximately 1% of AML cases (Refs Reference Tallman133, Reference Pagano134, Reference Barnard135).
DS-AMKL, non-DS-AMKL and adult AMKL are distinct in that they have different genetic abnormalities and different outcomes. DS-AMKL often presents by age 2 with a myelodysplastic phase that includes thrombocytopenia. Bone marrow aspiration is often difficult, with fibrosis detected on bone marrow biopsy (Refs Reference Hasle136, Reference Langebrake, Creutzig and Reinhardt137). The prognosis of DS-AMKL is favourable, with an approximately 80% cure rate (Refs Reference Creutzig138, Reference Rao139, Reference Gamis140). This outcome is based on the enhanced sensitivity of DS-AMKL blasts to chemotherapeutic drugs, especially cytarabine (ARA-C) (Ref. Reference Zwaan141). On the other hand, children with non-DS-AMKL fare worse, with a 5-year event free survival (EFS) of 22–28% (Refs Reference Barnard135, Reference Gamis142). The prognosis of adult AMKL is even worse, with a median survival of approximately 10 months (Ref. Reference Tallman133).
Several genes and pathways have been found in the aetiology of the different forms of AMKL. In DS-AMKL, trisomy 21 and somatic mutations in the haematopoietic transcription factor gene GATA1 are found in nearly every patient (Ref. Reference Wickrema and Crispino143). GATA1 mutations, which include short deletions, insertions and point mutations clustered within exon 2, are a key step in the pathogenesis of DS-AMKL (Ref. Reference Vyas and Crispino144). In every case, the mutations lead to a block in expression of the full-length protein, but allow for production of a shorter isoform, GATA-1s. Expression of GATA-1s alone is not sufficient to induce leukaemia, because humans with inherited mutant alleles fail to develop leukaemia (Ref. Reference Hollanda145). However, in mice, expression of GATA-1s in place of GATA-1 is sufficient to cause hyperproliferation of yolk sac and fetal liver MK progenitors (Ref. Reference Li146).
Distinct cytogenetic abnormalities, including t(1;Reference Eliades, Papadantonakis and Ravid22)(p13;q13), which leads to expression of the OTT-MAL fusion protein, t(9;Reference Adolfsson11), t(10;Reference Adolfsson11) and +8, are present in cases of non-DS paediatric AMKL (Ref. Reference Wickrema and Crispino143). As in the case of MPNs, alterations in tyrosine kinase signalling are probably necessary for AMKL. JAK3 mutations were originally identified in the DS-AMKL cell line, CMK, as well as in two DS-AMKL specimens, and were later detected in other DS-AMKL cases (Refs Reference Malinge, Izraeli and Crispino147, Reference Walters148, Reference Kiyoi149, Reference De Vita150, Reference Sato151, Reference Cornejo, Boggon and Mercher152). Apart from other rare mutations in c-MPL, c-KIT, JAK2 and FLT3, no other specific chromosomal rearrangements or genetic mutations have been identified in adult AMKL (Refs Reference Wickrema and Crispino143, Reference Malinge, Izraeli and Crispino147). Recent studies have shown that paediatric AMKL shows many more copy number alterations than other forms of AML (Ref. Reference Radtke153). However, how these changes influence the disease remains unknown. Rare instances of FLT3 mutations in AMKL do not influence prognosis (Ref. Reference Leow154). Recent studies have implicated dysregulation of miR-125b-2, which is overexpressed in AMKL relative to normal MKs, in DS-AMKL (Ref. Reference Klusmann155).
Two microarray studies comparing primary DS-AMKL versus non-DS AMKL specimens found 76 genes that discriminate between DS and non-DS AMKL. Genes encoding erythroid markers, such as glycophorin A and CD36, appear to be significantly overexpressed in DS-AMKL (Refs Reference Ge156, Reference Bourquin157). Further analysis of the gene expression data revealed that there is an overall increase in expression of chromosome 21 genes in DS-AMK than in non-DS AMKL (Ref. Reference Bourquin157). Forty-seven chromosome 21 genes were found to contribute to the enrichment by gene set enrichment analysis. However, the distinction between the two types of AMKL was not solely driven by differences in expression of chromosome 21 genes. The second study found that only 7 of 551 differentially expressed genes were on chromosome 21 (Ref. Reference Ge156).
Non-DS paediatric AMKL is characterised by an expansion of megakaryoblasts within the bone marrow and is frequently accompanied by hepatosplenomegaly, myelofibrosis and pancytopenia (Ref. Reference Carroll158, Reference Bernstein159, Reference Rubnitz160). The majority of cases are associated with the t(1;Reference Eliades, Papadantonakis and Ravid22) translocation, which results in fusion of the RNA-binding motif protein 15 (RBM15; aka OTT) and MKL1 (aka MAL) genes (Refs Reference Ma77, Reference Mercher161). The localisation and function of MAL, a coactivator of SRF with strong transcriptional activation properties, is deregulated by fusion with OTT (Refs Reference Miralles162, Reference Descot163). OTT is related to the SHARP transcription factor that acts in transcriptional repression complexes in canonical Notch signalling pathways (Refs Reference Ariyoshi and Schwabe164, Reference Oswald165). OTT might also inhibit myeloid differentiation (Ref. Reference Ma166). A conditional knock-in of the OTT-MAL fusion protein at its endogenous promoter resulted in deregulated transcriptional activity of the canonical Notch signalling pathway and abnormal fetal megakaryopoiesis. Cooperation between OTT-MAL and an activating mutation of MPL, but not the OTT-MAL knock-in alone, efficiently induced a short-latency highly penetrant AMKL that recapitulates the human phenotype (Ref. Reference Mercher167). Other cytogenetic abnormalities have been observed, including t(10;Reference Adolfsson11), which leads to CALM-AF10 fusion (Ref. Reference Abdelhaleem168), t(9;Reference Adolfsson11), +8 or +21 (Ref. Reference Barnard135).
Adult AMKL is a rare malignancy that comprises nearly 1% of adult AML cases (Refs Reference Tallman133, Reference Pagano134). Similar to the other subtypes of AMKL, adult AMKL is characterised by excessive production of immature megakaryoblasts within the bone marrow and extensive myelofibrosis. Peripheral blood features frequently include anaemia and thrombocytopenia. Adult AMKL frequently arises in individuals with an antecedent blood disorder or myelodysplastic syndrome (Ref. Reference Oki169). Although some patients achieve complete remission, the long-term outcome is significantly worse for adult AMKL than for other forms of adult AML, with a median survival of 40 weeks or less (Refs Reference Tallman133, Reference Pagano134, Reference Oki169).
To date, no specific chromosomal rearrangements or genetic mutations have been found in adult AMKL. In one study, it was noted that nearly 50% of patients had one or more cytogenetic abnormalities, including −5, −7, +8 or 11q involvement (Ref. Reference Oki169). These findings partially explain the poor outcome of this subtype of AMKL, although the poor prognosis is not fully dependent on cytogenetic abnormalities. Several studies have now reported that the tyrosine kinases JAK2 or JAK3 are mutated in a subset of AMKL patients, suggesting that perhaps JAK inhibitors should be considered. Given the poor prognosis for all patients with AMKL without DS, new, targeted therapies are desperately needed.
Summary and perspective
MKs have been recognised as rare marrow cells for nearly two centuries, but it was the elegant studies of Howell in 1890 and his coining of the term ‘MK’ that led to their broader appreciation as distinct entities. The field has entered a new age in which modern genetic and molecular biological tools are being applied to these unique cells both in situ and in vitro to tease out novel insights into their growth and differentiation. With this increasing knowledge base, it is likely that we will soon know the complete genetic defects related to abnormal megakaryopoiesis and will be in a strong position to develop new therapeutics for both excessive and inefficient megakaryopoiesis.
Acknowledgements
We thank Dr Sandeep Gurbuxani for a critical review and for providing the images used in Figure 2. We also thank the reviewers for their helpful suggestions. This review was supported in part by grants from the Samuel Waxman Cancer Research Foundation, the Leukemia and Lymphoma Society, and the National Cancer Institute (CA101774).


