Introduction
Fever-associated seizures or epilepsy (FASE) is primarily characterised by the occurrence of a seizure or epilepsy usually accompanied by a fever. It is common in infants and children generally in such forms as febrile seizures (FS), febrile seizures plus (FS+), Dravet syndrome (DS) and genetic epilepsy with febrile seizures plus (GEFSP). FS are the most common FASE occurring in neurologically normal infants and children between 3 months and 5 years, with an incidence of 2–5, 6–9 and 14–15% in the North American, Japanese and Pacific Islander populations, respectively (Refs Reference Deprez, Jansen and De Jonghe1, Reference Hedera2, Reference Tsuboi3, Reference Bird4, Reference Stanhope, Brody and Brink5). FS are accompanied by a fever but without neurological abnormalities, including encephalitis, developmental malformation, cerebral trauma and episodes of afebrile seizures (Ref. 6). FS are usually benign and resolve spontaneously without any antiepileptic drug therapy, following by no permanent neurological abnormality. The primary risk is a recurrence of FS. It is estimated that 33% of FS individuals will relapse. Fifty per cent of this group will experience another episode. Only 9% of FS individuals have more than three occurrences (Ref. Reference Johnson7). Although FS are isolated phenomena in most instances, it has also been reported that 2–7% of FS children may have subsequent afebrile and epileptic seizures in a later lifetime. This is 2–10 times as much as the general population (Ref. Reference Johnson7). In a population-based study, FS experiences are reported in 10–15% epilepsy cases (Ref. Reference Nabbout8). FS+ is characterised by FS attacked with fever continuing beyond 6 years or transferring to afebrile seizures. GEFSP is defined as a familial epilepsy syndrome with extremely variable expressivity. It includes classic FS, FS+ and generalised or localisation-related epilepsy accompanied by FS/FS+, such as absence seizures, myoclonic seizures, atonic seizures, myoclonic-astatic epilepsy, and more rarely, severe epileptic encephalopathy (Ref. Reference Poduri9). DS is commonly characterised by generalised tonic–clonic or hemiclonic epileptic seizures induced by fever in the first year of life, following by other seizure types, cognitive slowing or stagnation and behavioural abnormities in later childhood (Ref. Reference Lagae10).
Twin and family studies suggest that FASE is genetically predisposed. A positive family history is seen in 25–40% of FS children. The occurrence rate in siblings of patients with FS ranges from 9 to 22% (Ref. Reference Paul, Blaikley and Chinthapalli11). Several inheritance patterns have been described in FASE families, including polygenic, autosomal recessive and autosomal dominant with reduced penetrance (Refs Reference Deprez, Jansen and De Jonghe1, Reference Hedera2). Linkage analysis and mutation screening have identified several loci and disease-causing genes for FASE. The aetiology and the effect of fever in inducing seizures remain unclear.
This review provides an overview of the loci, the genes (Table 1), the associated molecular biology mechanisms and the fever-inducing effect of FASE (Fig. 1). This may improve our understanding of pathogenesis and may contribute to the clinical diagnosis of FASE.
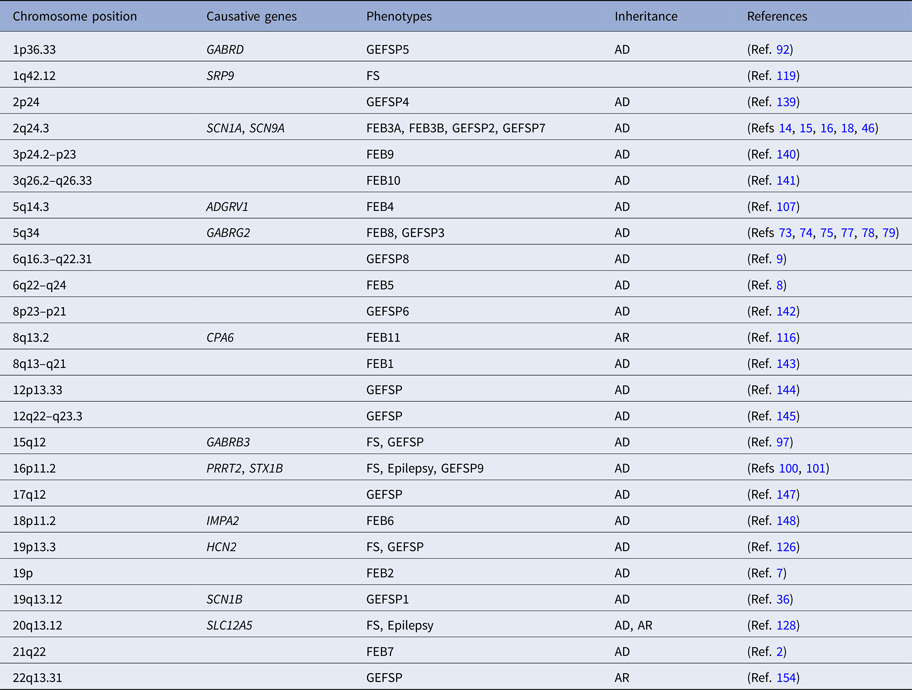
Table 1. The loci and genes associated with FASE
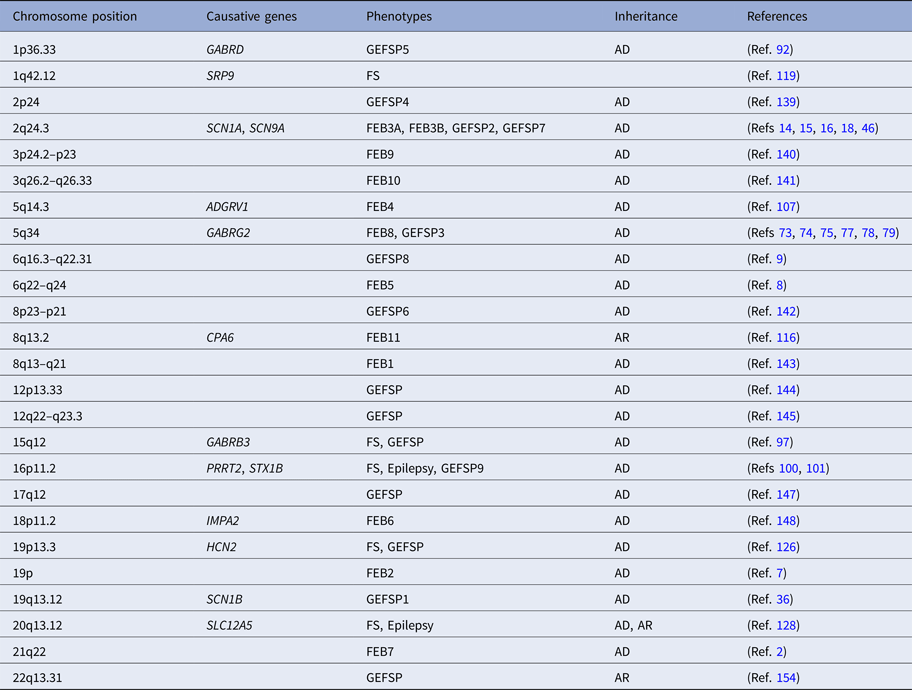
AD, autosomal dominant; ADGRV1, the adhesion G protein-coupled receptor V1 gene; AR, autosomal recessive; CPA6, the carboxypeptidase A6 gene; FASE, fever-associated seizures or epilepsy; FEB, familial febrile seizures; FS, febrile seizures; GABRB3, the γ-aminobutyric acid type A receptor β3-subunit gene; GABRD, the γ-aminobutyric acid receptor δ-subunit gene; GABRG2, the γ-aminobutyric acid A receptor γ2 gene; GEFSP, genetic epilepsy with febrile seizures plus; HCN2, the hyperpolarisation-activated cyclic nucleotide-gated potassium 2 gene; IMPA2, the myo-inositol monophosphatase 2 gene; PRRT2, the proline-rich transmembrane protein 2 gene; SCN1A, the sodium channel voltage-gated type I α-subunit gene; SCN1B, the sodium channel voltage-gated type I β-subunit gene; SCN9A, the sodium channel voltage-gated type IX α-subunit gene; SLC12A5, the solute carrier family 12 member 5 gene; SRP9, the signal recognition particle 9 kDa gene; STX1B, the syntaxin 1B gene.
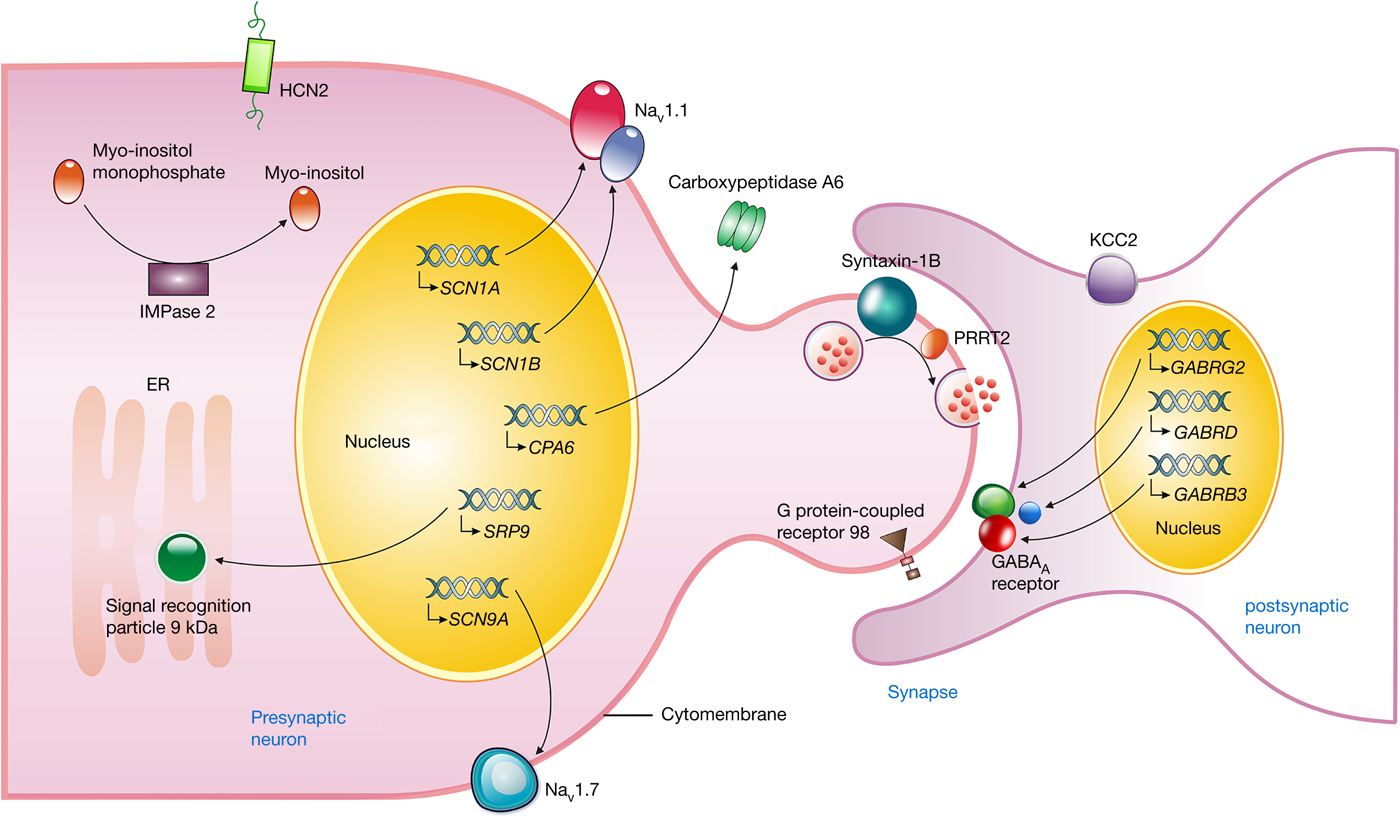
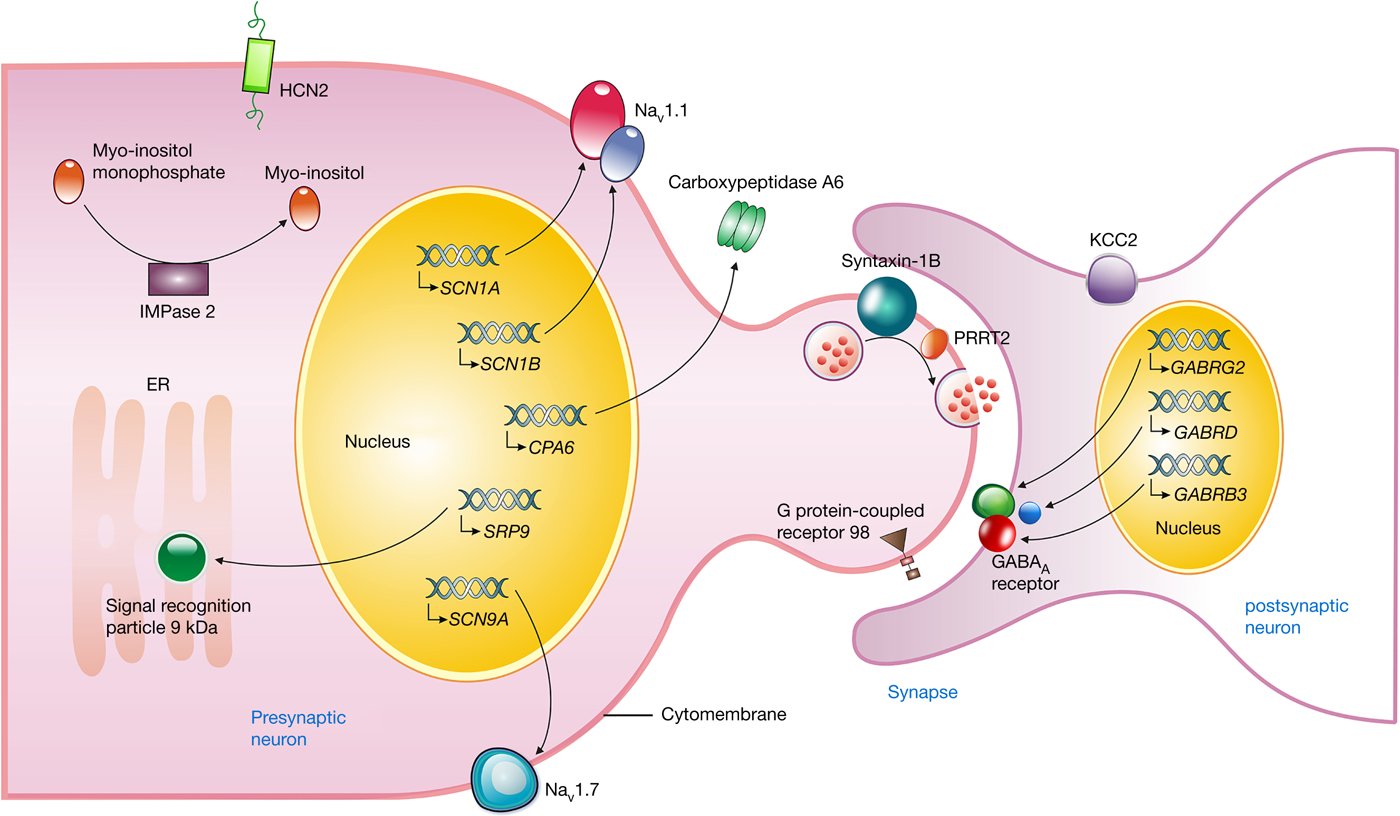
Figure 1. Neuronal location and proposed function of gene products associated with FASE. The SCN1A and SCN1B gene encode the α- and β-subunits of Nav1.1, respectively, which forms a fast inactivating voltage-dependent sodium channel and plays a critical role in the control of action potential generation and propagation in neurons. NaV1.7 encoded by the SCN9A gene may produce a rapidly activating and inactivating current with slowly repriming, which plays a role in pain mechanisms. The GABRG2, GABRD, and GABRB3 gene encode γ2, δ, and β3-subunit of the GABAA receptor, respectively, which mediates the majority of fast inhibitory synaptic transmission in the central nervous system. Syntaxin-1B, encoded by the STX1B gene, regulates synaptic vesicle fusion to the presynaptic membrane, as well as PRRT2, playing an important role in the presynaptic function and neuronal excitability. The mutant HCN2 channels enhance the current size in response to hyperpolarisation, and depolarise the membrane potential, taking the neuron closer to the firing potential. The myo-inositol monophosphatase (IMPase) 2, encoded by IMPA2 gene, catalyses the dephosphorylation of various myo-inositol monophosphates to free myo-inositol. The SRP9 variants interfere in targeting secretory proteins to the rough endoplasmic reticulum (ER) membrane. The GPR98 variants may affect neural cell–cell interactions or synapse structure. The mutant CPA6 protein affects the secretion into the extracellular matrix. KCC2, encoded by the SLC12A5 gene, establishes and maintains the hyperpolarising inhibitory postsynaptic potentials by conducting Cl− influx along its electrochemical gradient. FASE, fever-associated seizures or epilepsy; SCN1A, the sodium channel voltage-gated type I α-subunit gene; SCN1B, the sodium channel voltage-gated type I β-subunit gene; Nav1.1, the α-subunit of the neuronal voltage-gated sodium ion channel type 1; SCN9A, the sodium channel voltage-gated type IX α-subunit gene; GABRG2, γ-aminobutyric acid A receptor γ2 gene; GABRD, the γ-aminobutyric acid receptor δ-subunit gene; GABRB3, the γ-aminobutyric acid receptor type A β3-subunit gene; PRRT2, proline-rich transmembrane protein 2; STX1B, the syntaxin 1B gene; SLC12A5, the solute carrier family 12 member 5 gene; HCN2, the hyperpolarisation-activated cyclic nucleotide-gated potassium 2 gene; IMPA2, the myo-inositol monophosphatase 2 gene; SRP9, the signal recognition particle 9 kDa gene; GPR98, the G protein-coupled receptor 98 gene; CPA6, the carboxypeptidase A6 gene.
Disease-causing genes of FASE
The sodium channel voltage-gated type I α-subunit (SCN1A) gene
In 1999, Baulac et al. mapped the disease gene locus to 2q21–q33 within 22-cM between markers D2S156 and D2S2314 with a maximum pairwise logarithm of odds (LOD) score of 3.00 through linkage analysis of a pedigree with GEFSP. Moulard et al. confirmed linkage of GEFSP to chromosome 2q24–q33 in a different family (Refs Reference Baulac12, Reference Moulard13). Subsequently, heterozygous mutations, p.R1648H and p.T875M, in the SCN1A gene were identified in the two pedigrees described by Baulac et al. and Moulard et al., respectively (Ref. Reference Escayg14). In 2005, a heterozygous mutation, p.M145T, in the SCN1A gene was identified in an Italian pedigree with familial febrile seizures (FEB) (Ref. Reference Mantegazza15). The phenotypes in this pedigree were first designated FEB3A. Because three individuals developed afebrile partial seizures, partial complex seizures or nocturnal secondary generalised tonic–clonic seizures, the FEB3A was best classified as GEFSP2. There were at least 786 SCN1A gene mutations identified without specific mutational hotspots, which were responsible for a kind of seizure-related disorders, including FS, GEFSP, severe myoclonic epilepsy borderline (SMEB) and DS (Refs Reference Zuberi16, Reference Parihar and Ganesh17). The SCN1A mutations are de novo in 88% probands, and only 12% mutations inherited from one of the parents (Ref. Reference Parihar and Ganesh17). SCN1A mutations were responsible for 21.2% of GEFSP individuals and accounted for 70–80% of DS (Refs Reference Orrico18, Reference Mulley19). The missense or inherited mutations occur more frequently in FS and GEFSP patients, whereas de novo mutations frequently result in SMEB and DS (Ref. Reference Parihar and Ganesh17).
The human SCN1A gene, mapped to 2q24.3, spans ~159.8 kb. It contains 28 exons and encodes the α-subunit of the neuronal voltage-gated sodium ion channel type 1 (Nav1.1). The Nav1.1 channel, identified in peripheral nervous systems and cardiac myocytes, as well as central nervous systems (CNS) (Refs Reference Lossin20, Reference Catterall21, Reference Isom, De Jongh and Catterall22), is an integral heteromeric membrane protein which forms a fast inactivating voltage-dependent sodium channel and plays a critical role in controlling the generating and propagating of action potential in excitable cells, such as neurons (Ref. Reference Deng, Xiu and Song23). The Nav1.1 channel consists of an α-subunit and two smaller auxiliary β-subunits (Ref. Reference Meisler and Kearney24). The α-subunit, a large 260 kDa transmembrane protein including a voltage transducer and a sodium-passing unit, regulates the sodium ion trafficking and may work alone as a channel. The Scn1a expression in the CNS is detected postpartum in rodents and elevates up to adulthood, especially in dendrites, cell bodies and the parvalbumin-positive inter-neuronal axon initial segments (Refs Reference Ogiwara25, Reference Gong26, Reference Westenbroek, Merrick and Catterall27, Reference Duflocq28). SCN1A mutations inactivate the channel and result in persistent inward sodium current. This gain-of-function mutation may increase neuronal excitability by overlong membrane depolarisation, which is a potential pathogenesis of GEFSP (Ref. Reference Lossin29).
Scn1a −/− mice exhibited serious ataxia and epileptic seizures, and survived for only 15 days postpartum, while heterozygous mutation mice presented with spontaneous seizures, living for 21 days or longer postpartum (Refs Reference Ogiwara25, Reference Yu30). Scn1a deficiency in inhibitory interneurons, especially in GABAergic interneurons, effected sodium ion currents and evoked action potentials, which resulted in seizures and premature death (Refs Reference Parihar and Ganesh17, Reference Cheah31, Reference Martin32, Reference Han33). Interneuron dysfunction appears to play a crucial role in the pathogenic mechanism of Scn1a-related seizure disorders (Refs Reference Ogiwara25, Reference Yu30).
The sodium channel voltage-gated type I β-subunit (SCN1B) gene
The SCN1B gene mutations lead to GEFSP, very rarely DS and cardiac conduction diseases (Ref. Reference Bouza and Isom34). In 1998, Wallace et al. mapped the disease locus on 19q13.1 in a large family with GEFSP and first detected a heterozygous p.C121W mutation in the SCN1B gene (Ref. Reference Wallace35). Subsequently, Wallace et al. identified the pathogenic mutations in another GEFSP family (Ref. Reference Wallace36), and two DS individuals harbouring pathogenic homozygous SCN1B variants have been reported (Ref. Reference Steel37). Heterozygous SCN1B mutations result in the milder epilepsy syndrome known as GEFSP with variable penetrance (Refs Reference Wallace35, Reference Steel37). Homozygous SCN1B mutations may cause classical more severe DS (Refs Reference Ogiwara38, Reference Patino39).
The human SCN1B gene is mapped to 19q13.12, spanning approximately 9.7 kb. It contains five exons and encodes the auxiliary β-subunit of the Nav1.1. The 33–36 kDa auxiliary β-subunit, including a large N-terminal extracellular domain and a short C-terminal intracellular domain, may regulate channel expression, transfer the channel protein to the cell membrane, and modulate channel-gating through interacting with the cytoskeleton and other proteins (Refs Reference Parihar and Ganesh17, Reference Deng, Xiu and Song23, Reference Williams and Battaglia40).
The Scn1b-null mice may be DS models (Ref. Reference Ramadan41). Homozygous Scn1b mutant mice may have an increased action potential firing rate in mutant subicular and layer 2/3 pyramidal neurons with no change in firing or synaptic properties of GABAergic interneurons, which differs from the Scn1a-based DS models (Ref. Reference Reid42). The neurons in Scn1b C121W/+ mice are more excitable and sensitive to the temperature, which leads to a lowered threshold for action potential triggering (Ref. Reference Hatch, Reid and Petrou43). The effects of antiepileptic drugs, including carbamazepine and retigabine, are enhanced in slices from the Scn1b (p.C121W) heterozygous mice with decreased neuron excitability (Ref. Reference Hatch, Reid and Petrou43). Knockdown of Scn1b in mouse decreases I(A) channel densities in isolated cortical neurons, prolongs action potential waveforms and increases repetitive firing in cortical pyramidal neurons (Ref. Reference Marionneau44).
The sodium channel voltage-gated type IX α-subunit (SCN9A) gene
In 1999, the disease gene locus was mapped to 2q23–q24 within the 10-cM interval with LOD score of 8.08 at D2S2330 in a large Utah family with 11 members affected by FS-designated FEB3B (Ref. Reference Peiffer45). Ten additional members, including one DS individual, later developed afebrile seizures (Ref. Reference Singh46). FEB3B was named later to be GEFSP7 as the phenotype most consistent with GEFSP. Subsequently, a heterozygous mutation, p.N641Y, was identified in the SCN9A gene in this pedigree. Additionally, nine missense SCN9A variants, all in highly conserved amino acids, were identified in 109 patients with sporadic DS, although six patients in this group also had SCN1A mutations (Ref. Reference Singh46). Rare SCN9A variants occurred in 12% of the DS population, significantly higher than 2.2% in healthy controls (Refs Reference Mulley19, Reference Steel37).
The human SCN9A gene, mapped to 2q24.3, spans approximately 180.6 kb. It contains 27 exons and encodes Nav1.7 subunit. It is a peripheral nervous system channel and primarily expressed in the dorsal root ganglia (DRG) neurons, sympathetic ganglion neurons (Refs Reference Toledo-Aral47, Reference Rush48), myenteric neurons (Ref. Reference Sage49), olfactory sensory neurons (Ref. Reference Ahn50), visceral sensory neurons (Ref. Reference Muroi51), smooth myocytes (Refs Reference Holm52, Reference Jo53, Reference Saleh54) and their small-diameter axons (Ref. Reference Persson55). Nav1.7 is also expressed in a number of nonexcitable cells, including prostate tumour cells, breast tumour cells (Refs Reference Diss56, Reference Fraser57), erythroid progenitor cells and immune cells (Refs Reference Hoffman58, Reference Kis-Toth59).
Nav1.7, a threshold channel, may produce a rapid activation and inactivation current with slow recovering from inactivation (Refs Reference Klugbauer60, Reference Rush, Cummins and Waxman61). The slow closed-state inactivation permits the channel to produce a significant ramp current and results in small, slow depolarisation, which promotes subthreshold stimuli to increase the feasibility of the neurons exceeding the threshold and generating action potentials (Refs Reference Cummins, Howe and Waxman62, Reference Herzog63). Nav1.7 in DRG neurons and nociceptors takes part in physiological processes of pain, particularly in the generation of inflammatory pain (Refs Reference Hayes and Wang64, Reference Dib-Hajj65, Reference Djouhri66). Gain-of-function SCN9A mutations may decrease DRG neurons threshold and lead to severe neuropathic pain, such as inherited erythromelalgia (Refs Reference Dib-Hajj65, Reference Waxman67, Reference Mansouri68). Loss-of-function SCN9A mutations result in an indifference to pain, such as congenital insensitivity to pain (Refs Reference Waxman67, Reference Mansouri68). Other SCN9A mutations, reported to be associated with paroxysmal extreme pain disorder, impair Nav1.7 channel inactivation and produce hyperexcitability in DRG neurons (Refs Reference Waxman67, Reference Mansouri68). Additional studies have linked Nav1.7 to other kinds of sense, such as olfactory sensation (Refs Reference Ahn50, Reference Weiss69), the afferent nerve fibre of the cough reflex (Ref. Reference Muroi51), acid-sensing (Ref. Reference Smith70) and paroxysmal itch (Ref. Reference Devigili71).
Homozygous Scn9a N641Y/N641Y knock-in mice have a remarkable lower threshold to clonic and tonic–clonic seizures associated with electrical stimulation, compared to wild-type mice (Ref. Reference Singh46). However, no measurable Nav1.7 expression has been identified in the CNS (Refs Reference Toledo-Aral47, Reference Felts72), and no patients with other SCN9A mutations associated with the extreme pain or insensitivity to pain disorders have been reported to experience seizures. The contribution of SCN9A mutations to epilepsy is not well understood.
The γ-aminobutyric acid A receptor γ2 (GABRG2) gene
In 2001, Wallace et al. detected a heterozygous mutation, p.R43Q, in the GABRG2 in a four-generation family with FS and childhood absence epilepsy (Ref. Reference Wallace73). Subsequently, Kananura et al. identified the GABRG2 IVS6 + 2 T > G mutation in a German family with FS and childhood absence epilepsy (Ref. Reference Kananura74). The GABRG2 mutations caused a spectrum of seizure disorders, ranging from FS and GEFSP to DS in unrelated families (Refs Reference Baulac75, Reference Carvill76, Reference Audenaert77, Reference Harkin78, Reference Sun79, Reference Todd80, Reference Le81).
The human GABRG2 gene is mapped to 5q34, spanning ~87.8 kb. It contains 11 exons and encodes the γ-aminobutyric acid receptor type A (GABAA receptor) γ2-subunit. GABAA receptors are ligand-gated heteropentameric chloride ion channels and highly expressed in brain, including cerebellum, cerebral cortex, medulla, occipital pole, frontal lobe, temporal lobe and putamen, taking part in the fast inhibitory synaptic transmission in the mammalian CNS (Refs Reference Kang, Shen and Macdonald82, Reference Hirose83). GABAA receptors are combinations of two α-subunits, two β-subunits and one γ2-subunit or δ-subunit. The γ2-subunit is crucial for GABAA receptor trafficking, assembling and synaptic maintaining (Ref. Reference Kang, Shen and Macdonald82). The GABRG2 mutations may decrease surface expression γ2-subunit and inhibitory synaptic strength in neuron through misfolding, abnormal assembly, endoplasmic reticulum (ER) retention, enhanced degradation or rapid endocytosis. This may be exacerbated by fever and result in FS or lower the threshold of afebrile seizures (Ref. Reference Kang, Shen and Macdonald82). There is also evidence that lower temperatures increase GABAA receptor stability (Ref. Reference Huang84).
The p.R43Q mutation reduced α1β2γ2 receptor surface expression by the abnormal assembly, ER retention and degradation (Ref. Reference Huang84). The p.R177G mutation reduced GABA-evoked whole-cell current amplitudes and resulted in smaller peak current densities by reducing cell surface expression of α1β2γ2L receptors. This potentially reduces GABAergic phasic inhibition and lowers the seizure threshold by elevating neuronal hyperexcitability (Ref. Reference Todd80). The p.K289M mutation may reduce miniature inhibitory postsynaptic currents duration by promoting its deactivation by impairing the stability of the channel open state, and the p.Q351* mutation may prevent the cell surface trafficking of both α1β2γ2 and α1β2 receptors (Refs Reference Harkin78, Reference Eugene85, Reference Bianchi86, Reference Hales87, Reference Kang, Shen and Macdonald88). Heterozygous knock-in mice carrying p.R43Q mutation showed spontaneous spike-wave discharges and thermal-induced seizures at a critical time during development (Refs Reference Reid89, Reference Tan90, Reference Chiu91).
The γ-aminobutyric acid receptor δ-subunit (GABRD) gene
In 2004, a heterozygous p.E177A mutation in the GABRD gene was reported in affected individuals of a GEFSP pedigree. The alteration resulted in decreased GABAA receptor current amplitudes (Ref. Reference Dibbens92).
The human GABRD gene was mapped to 1p36.33. The 1A and 1B variants contain nine exons and the 1C variant contains eight exons. The GABRD gene encodes the δ-subunit of the GABAA receptor (Ref. Reference Windpassinger93). The δ-subunit is affinitive to GABA and is the preferential target of neuroactive steroids (Ref. Reference Feng94). The heterozygous or homozygous p.E177A mutation in GABRD gene decreased macroscopic current amplitudes of α1β2δ receptors through a shortened mean channel open time (Refs Reference Feng95, Reference Macdonald, Kang and Gallagher96).
The γ-aminobutyric acid type A receptor β3-subunit (GABRB3) gene
The mutations in human GABRB3 gene are associated with a broad phenotypic spectrum which includes FS, GEFSP, epilepsy with myoclonic-atonic seizures and early-onset epileptic encephalopathies (Ref. Reference Moller97). Recently, a novel de novo heterozygous missense variant c.695G>A in the GABRB3 gene was identified in a DS individual (Ref. Reference Le81).
The GABRB3 gene, mapped to 15q12 and spanning ~250 kb, contains 10 exons and encodes β3-subunit of the GABAA receptor. The β3-subunit includes a large extracellular NH2-terminal domain, four hydrophobic transmembrane domains (M1–M4), an M3–M4 intracellular loop, and a COOH-terminus (Ref. Reference Le81). The GABRB3 mutations result in reduced GABA-induced current amplitudes or GABA sensitivity. GABAergic disinhibition caused by reduced receptor function is a major mechanism for epilepsy (Refs Reference Le81, Reference Moller97).
The proline-rich transmembrane protein 2 (PRRT2) gene
Mutations in the PRRT2 gene were responsible for 39.4–65% of episodic kinesigenic dyskinesia (EKD) (Ref. Reference Meneret98), 83% of familial infantile convulsions with paroxysmal choreoathetosis (ICCA) (Ref. Reference Heron99) and 82% of benign familial infantile seizures (BFIS) (Ref. Reference Heron99). In 2016, a duplication mutation PRRT2 p.R217Pfs*8 was identified in our Chinese family with FS (Ref. Reference Zheng100). Mutations in the PRRT2 gene were identified in 18.4% (25/136) epileptic patients with FS (Ref. Reference He101). Up to date, at least 40 mutations have been noted in the literature, and EKD, ICCA and BFIS are main phenotypes of disorders related to these mutations.
The human PRRT2 gene, mapped to 16p11.2, spans approximately 315 kb. It contains four exons and encodes a protein with 340 amino acids. PRRT2 is widely expressed in the brain, including the cerebral cortex, hippocampus and cerebellum. The p.R217Pfs*8 mutation may result in a truncated protein lacking membrane targeting and being mislocated in the cytoplasm, which is predicted to influence the function of specific types of ion channels (Ref. Reference Chen102). Truncated p.R217Pfs*8 protein was also found to lose its ability to interact with the synaptosomal-associated protein 25-KD (SNAP25; MIM 600322), which is a presynaptic plasma-membrane-bound protein and plays a role in neurotransmitter release from synaptic vesicles (SVs) (Ref. Reference Heron99). The functional abnormality of specific types of ion channels and abnormal neurotransmitter release may thereby give rise to seizures (Ref. Reference Zheng100).
The syntaxin 1B (STX1B) gene
In 2014, p.G56* mutation and a complex mutation (p.Lys45delinsArgMetCysIleGlu and p.Leu46Met) in the STX1B gene were identified in two families with FS and epilepsy in early childhood (Ref. Reference Schubert103). STX1B mutations may lead to a wide phenotypic spectrum, including FS, GEFSP and epileptic encephalopathies (Ref. Reference Schubert103).
The human STX1B gene encodes syntaxin-1B, a highly conserved isoform of syntaxin-1, as well as syntaxin-1A. Syntaxin-1B plays an important role in the presynaptic function and neuronal excitability. Together with synaptobrevin and SNAP25, syntaxin-1 composes the soluble N-ethylmaleimide-sensitive factor attachment receptor complex, which regulates SV fusion to the presynaptic membrane (Ref. Reference Schubert103). Syntaxin-1A and -1B can partially replace each other, but syntaxin-1B seems to be more important with broader expression pattern (Ref. Reference Foletti104). It was also reported that syntaxin-1B modulates γ-aminobutyric acid and glutamate release (Ref. Reference Bragina105). Knockdown of stx1b in zebrafish larvae displayed seizure-like behaviour and epileptic discharges, including polyspiking discharges and high-frequency oscillations, which were significantly sensitive to elevated temperature (Ref. Reference Schubert103). Haploinsufficiency for STX1B is a shared mechanism of pathogenesis in FS and epilepsy, and other unknown factors can modify the phenotype (Ref. Reference Schubert103).
The adhesion G protein-coupled receptor V1 (ADGRV1) gene
The human ADGRV1 gene also is named the G protein-coupled receptor 98 gene (GPR98), the monogenic audiogenic seizure susceptibility 1 gene (MASS1) or the very large G protein-coupled receptor 1 gene (VLGR1).
In 2000, Nakayama et al. identified a disease gene locus of FS on 5q14–q15 through linkage analysis of a large FS pedigree. Subsequent analysis showed significant linkage disequilibria with FS presenting in 47 Japanese families by transmission disequilibrium tests (Ref. Reference Nakayama106). Subsequently, p.S2652* mutation in the GPR98 gene, causing loss-of-function due to a deletion of C-terminal 126 amino-acid, was identified in a family with FS and afebrile seizures (Ref. Reference Nakayama107). In 2006, by genome-wide scan in a multi-generational family with FS and epilepsy, Deprez et al. identified a disease gene locus on 5q14.3–q23.1 covering the FEB4 locus with a maximal multipoint LOD score of 3.12, but no disease-causing mutation was detected in the ADGRV1 gene (Ref. Reference Deprez108). ADGRV1 gene mutations are generally associated with Usher syndrome 2 and present with congenital sensorineural hearing loss and progressive retinitis pigmentosa, respectively leading to deafness and blindness.
The human ADGRV1 gene, mapped to 5q14.3, spans approximately 605.5 kb. It contains 90 exons (Ref. Reference McMillan and White109). The expression of ADGRV1 is low in most mature tissues (Ref. Reference Nikkila110), but is significant in embryonic CNS, particularly in the ventricular zone (Refs Reference McMillan111, Reference McMillan112, Reference McMillan and White113). The big ectodomain of ADGRV1 comprises a number of repeated motifs, including 35 calcium binding, Calx-β repeats, and seven copies of epitempin repeat (Refs Reference McMillan and White109, Reference Scheel, Tomiuk and Hofmann114). ADGRV1 mutations may affect neural cell–cell interactions or change synaptic structure in specific regions of the CNS, resulting in seizures susceptibility (Ref. Reference McMillan and White113).
Frings mouse is an autosomal recessive inherited animal model for epilepsy evoked by auditory stimuli. In 2001, the Adgrv1 p.V2250* mutation, which led to the transmembrane and cytoplasmic domains of ADGRV1 losing, was identified in Frings mouse. The abnormality disrupts the development of signal regulating neuron and up-regulates susceptibility to audiogenic seizures (Ref. Reference Skradski115).
The carboxypeptidase A6 (CPA6) gene
In 2012, using a genome-wide autozygosity mapping followed by candidate gene analysis of a consanguineous Moroccan family with FS, FEB11 was located in a 9.6 Mb region on chromosome 8q12.1–q13.2. A homozygous mutation, p.A270V, in the CPA6 gene was identified in this pedigree (Ref. Reference Salzmann116).
The human CPA6 gene, mapped to 8q13.2, spans approximately 324 kb. It contains 11 exons and encodes a carboxypeptidase, which removes amino acids from the C-termini of proteins by hydrolysis. In mice, the protein is expressed in brain and takes part in neuropeptide cleavage (Ref. Reference Belhedi117). Carboxypeptidases may be involved in the biosynthesis of neuroendocrine peptides. CPA6 contains a pro region consisting of about 90 amino acids, which assists the folding of the active carboxypeptidase domain and the cleavage of the pro region activating the enzyme (Ref. Reference Wei118). The mutant protein has decreased activity due to impaired secretion into the extracellular matrix. For example, the p.A270V homozygous mutation has about 40% residual enzyme activity which is consistent with loss of function (Ref. Reference Salzmann116). The CPA6 protein may cleave angiotensin I to angiotensin II and take part in brain development and neuronal migration. It also inactivates neurotensin, which takes part in thermal regulation, stress and depression (Ref. Reference Belhedi117). Additionally, four rare missense mutations and a common marker in CPA6 gene, putatively affecting enzymatic activity and protein levels, have been reported to be related to FS (Ref. Reference Belhedi117).
The signal recognition particle 9 kDa (SRP9) gene
In 2014, a quantitative trait locus for FS susceptibility was mapped on chromosome 1, and the Srp9 gene was identified as an FS susceptibility gene in mice (Ref. Reference Hessel119). The SRP9 expression in mesial temporal lobe epilepsy (mTLE) patients with FS is higher than mTLE patients without FS, and SRP9 promoter SNP rs12403575 was found to be associated with mTLE and FS (Ref. Reference Hessel119).
SRP9 is a subunit of SRP, a cytoplasmic ribonucleoprotein complex with at least three distinct functions, including signal recognition, translational arrest and targeting nascent membrane proteins to the rough ER (Refs Reference Siegel and Walter120, Reference Lingelbach121). SRP comprises two functional domains, including the S-domain (SRP19, 54, 68 and 72) and the ALU domain (7SL RNA and the SRP9/SRP14 heterodimer) (Refs Reference Walter and Blobel122, Reference Bovia, Bui and Strub123). Reduced SRP9 levels result in susceptibility to FS by interfering with the expression of membrane proteins targeted to the ER, such as glutamate receptors (Refs Reference Hessel119, Reference Lakkaraju124, Reference Lakkaraju125).
The hyperpolarisation-activated cyclic nucleotide-gated potassium 2 (HCN2) gene
In 2010, a delPPP variant in the HCN2 gene was identified in 2.5% patients with FS and in 2.3% patients with GEFSP, while the deletion was present in only 0.2% controls and was not identified in cases with idiopathic generalised epilepsy, but without FS. The current size of delPPP variant elevated 35% in the course of hyperpolarisation, and the current change may depolarise the membrane potential, triggering the action potential and thus raise neuronal excitability (Ref. Reference Dibbens126).
In 2013, a heterozygous p.S126L mutation in HCN2 was observed in two unrelated FS patients. The mutant channels resulted in the important cAMP-independent enhanced availability of I h (a hyperpolarisation-activated cation current) during high temperatures, which leads to hyperthermia-induced neuronal hyperexcitability and elevates sensitivity to temperature in some individuals with FS (Ref. Reference Nakamura127).
The solute carrier family 12 member 5 (SLC12A5) gene
In 2014, a missense mutation p.R952H in the SLC12A5 gene, leading to impairment of neuronal Cl− extrusion and cortical dendritic spine formation, was identified in an Australian family with early childhood onset of FS (Ref. Reference Puskarjov128).
The human SLC12A5 gene, mapped to 20q13.12, encodes the neuron-specific K-Cl co-transporter KCC2 protein. Mouse KCC2 protein was divided into KCC2a and KCC2b, differing in the N-terminal. Although the two isoforms coexist in the brain with similar distribution in the neonatal CNS of mice, the KCC2b expression is significantly increased during the following brain development and accounts for >90% of the total KCC2 protein in the adult murine brain (Refs Reference Uvarov129, Reference Khirug130, Reference Stein131). KCC2 is crucial in establishing and maintaining the hyperpolarising inhibitory postsynaptic potentials by conducting Cl− influx along its electrochemical gradient, which conveys the GABAergic inhibition in the mammalian brain (Refs Reference Blaesse132, Reference Chamma133, Reference Chamma134). Independently of an ion transporter, rodent KCC2 takes part in the morphological and functional maturation of cortical dendritic spines as an important structural protein (Refs Reference Li135, Reference Fiumelli136). The SLC12A5 mutations may lead to disbalance of GABAergic and glutamatergic transmitter systems, and generate seizures further (Ref. Reference Puskarjov128).
Mutations in the Drosophila melanogaster kcc gene, a homologue of human SLC12A5, were reported to increase seizure susceptibility (Ref. Reference Hekmat-Scafe137). Homozygous Slc12a5 knockout mice may develop generalised seizures and die during the second postnatal week. While heterozygous knockout mice, expressing ~50% of the wild-type KCC2 protein, were subject to pentylenetetrazole-induced seizures (Ref. Reference Woo138).
Loci without identified disease genes
2p24
In 2005, a disease locus was mapped to a 3.24 cM region on 2p24 with maximum two-point LOD score of 4.22 at marker D2S305 by genome-wide linkage analysis and haplotype analysis of a four-generation Belgian family with FS and GEFSP. The candidate region was reduced to a 2.14 cM interval based upon analysis of additional 50 nuclear and multiplex families with FS and epilepsy (Ref. Reference Audenaert139), but no gene mutation was identified.
3p24.2–p23
In 2007, a disease locus was mapped on chromosome 3p24.2–p23 within a 9.4 cM interval between markers D3S2336 and D3S3518 by genome-wide linkage and haplotype analysis of a four-generation French family in which 13 members had FS, and five of whom subsequently developed childhood absence epilepsy, but no disease-causing mutation was identified (Ref. Reference Nabbout140).
3q26.2–q26.33
In 2008, Dai et al. detected a 10.7 Mb on 3q26.2–q26.33 between markers D3S3656 and D3S1232 with a maximum multipoint LOD score of 5.27 at D3S1565 by linkage analysis of a four-generation Chinese family with FS, but no disease-causing mutation was identified (Ref. Reference Dai141).
6q16.3–q22.31
By genome-wide analysis of a four-generation family with GEFSP, Poduri et al. identified a disease locus on chromosome 6q16.3–q22.31 within 12.4 cM region between D6S962 and D6S287 with maximum multipoint LOD score of 4.68. However, no deletions or duplications were identified by copy number assessment, and the causative mutation was excluded in 16 candidate genes (Ref. Reference Poduri9).
6q22–q24
In 2002, Nabbout et al. identified a 6.4 cM on 6q22–q24 between D6S1620 and D6S975 with maximum multipoint LOD score of 4.82 by analysis of two families with FS, and excluded mutations in five candidate genes, including the A-kinase anchoring protein 18 gene, the syntaxin seven gene, the putative neurotransmitter receptor gene, the G protein receptor 57 gene, and the G protein receptor 58 gene (Ref. Reference Nabbout8).
8p23–p21
In 2008, a disease locus was mapped to a 7.3 Mb region on chromosome 8p23–p21 between D8S1706 and D8S549 by analysis of two French families with GEFSP. Sequence analysis excluded mutations in the coding exons of several candidate genes, including the myotubularin related protein 9 gene (MTMR9), the myotubularin related protein 7 gene (MTMR7), the cathepsin B gene (CTSB), the sarcoglycan zeta gene (SGCZ) and the ATPase H+ transporting V1 subunit B2 gene (ATP6V1B2). No ion channel genes are located in the critical region (Ref. Reference Baulac142).
8q13–q21
In 1996, the first disease gene locus was mapped to 8q13–q21 between D8S553 and D8S279 with multipoint LOD score of 3.40 in an FEB family (Ref. Reference Wallace143). In 2012, the researchers excluded mutations in the CPA6 gene within this region in this family, but could not exclude the possibility of a mutation in the promoter or noncoding region or a copy number variant (Ref. Reference Salzmann116).
12p13.33
In 2011, Morar et al. identified a disease locus on chromosome 12p13.33 with LOD score of 3.84 by analysis of a large consanguineous family with GEFSP from an endogamous Roma/Gypsy sub-isolate. Sequencing of the coding regions of three genes linked to neurotransmitter transport and release on 12p did not identify a causative mutation (Ref. Reference Morar144).
12q22–q23.3
In 2004, a disease gene locus was mapped to 12q22–q23.3 within a 10.35 cM interval between markers D12S101 and D12S360 by genome-wide scan and linkage analysis of a five generation pedigree with familial temporal lobe epilepsy and FS by Claes et al. (Ref. Reference Claes145). Given that the combination of FS, FS+ and partial seizures epilepsy is now regarded as GEFSP, the diagnosis of pedigree described by Claes et al. was suggested to GEFSP. In 2007, the disease gene locus of a North American Caucasian family with seven affected individuals with FS was mapped to 12q22–q23.3 within 60 cM between markers D12S297 and D12S2070. Three of seven patients also had afebrile seizures during childhood (Ref. Reference Gurnett146). There are more than 50 genes within the overlapping interval, and resequencing did not identify mutations in some genes including the vesicular glutamate transporter 3 gene (VGLUT3), the achaete-scute family bHLH transcription factor 1 gene (ASCL1), the ADP ribosylation factor like GTPase 1 gene (ARL1), the transmembrane protein 16D gene (TMEM16D) (Ref. Reference Gurnett146).
17q12
In 2013, Hardies et al. found a 17q12 duplication cosegregating in a four-generation GEFSP family (Ref. Reference Hardies147). The 17q12 duplication had previously been described in individuals with neurodevelopmental disorders, such as intellectual disability and autism spectrum disorder. In some patients, other features, such as oesophageal atresia and dysmorphic facial features are also described. Seizures were described in four of 43 reported individuals with the duplication and four of 55 patients with the deletion, which indicated that the express alternation or mutation of a certain located in 17q12 may cause GEFSP (Ref. Reference Hardies147).
18p11.2
In 2004, by genome-wide linkage analysis of 48 Japanese families in which at least two children were affected with FS, Nakayama et al. identified a 2.4 Mb region on 18p11.2 with maximum nonparametric LOD score of 3.68 at D18S1158, and suggested an association of a common haplotype in the myo-inositol monophosphatase 2 gene (IMPA2) with FS by only haplotype analysis (Ref. Reference Nakayama148). In another study, no mutations in the IMPA2 gene were detected in 96 unrelated Caucasian individuals with FS (Ref. Reference Blair149).
The IMPA2 gene contains eight exons and encodes myo-inositol monophosphatase (IMPase) 2 (Ref. Reference Yoshikawa150). The IMPase plays a crucial role in the phosphatidylinositol signalling pathway. It catalyses the dephosphorylation of many myo-inositol monophosphates to free myo-inositol (Ref. Reference Berridge and Irvine151). The variants of IMPA2 are associated with bipolar disorders (Refs Reference Sjoholt152, Reference Sjoholt153). IMPase 2 may be inhibited by lithium by decreasing seizure threshold in bipolar disorders therapy and may be stimulated by an anticonvulsant carbamazepine (Refs Reference Nakayama148, Reference Blair149). However, the real role of the IMPA2 gene mutation in FS should be further studied.
19p
In 1998, by linkage analysis of a large FS family from American Midwest, the disease gene locus was mapped to 19p within an 11.7 cM between D19S591 and D19S395 with a maximum pairwise LOD score of 4.52 (Ref. Reference Johnson7). No mutations have been identified in a lot of possible candidate genes within this region, including the G protein subunit alpha 11 gene (GNA11), the G protein subunit alpha 15 gene (GNA15), the thromboxane A2 receptor gene (TBXA2R), the zinc finger protein 57 gene (ZNF57) and the megakaryocyte-associated tyrosine kinase gene (MATK) (Ref. Reference Johnson7).
21q22
In 2006, Hedera et al. described a 6.5 cM region between D21S1909 and D21S1444 on 21q22 with maximum multipoint LOD score of 3.62 at D21S1910 by linkage analysis of a five-generation FS family, and excluded mutations in four ion-channel genes, including SCN1B, SCN1A, the sodium channel voltage-gated type II β-subunit gene (SCN2B) and the GABRG2 gene (Ref. Reference Hedera2). Additionally, the pedigree was suggested to be GEFSP due to two family members with recurrent FS coexisting with afebrile generalised tonic–clonic seizures at age of 2–3 years.
22q13.31
In 2013, a disease gene locus was narrowed to a 527 kb interval on chromosome 22q13.31 between 18GTchr22 and 15ACchr22b with a maximum multipoint LOD score of 2.51 by analysis of a consanguineous Tunisian family with three GEFSP patients. Whole-exome sequencing identified no mutation. The mutation may have been missed or a disease-associated copy number variant may exist in the pedigree (Ref. Reference Belhedi154).
Effects of increased temperature on seizures
Mutations in ion channel genes may result in hyperexcitability of a neuron through enhancing excitatory or decreasing inhibitory neurotransmission. Many mutations in ion channel genes or GABAA receptor subunit genes have been identified in FASE patients. However, what is not well understood how fever generates FS.
Hippocampal neurons, in vitro, can exhibit significant increases in intrinsic excitability when exposed to hyperthermic conditions (Ref. Reference Kim and Connors155). L-type voltage-gated calcium channels are fairly active at hyperpolarised potentials and drive intrinsic firing at temperatures ≥37 °C, which may contribute to a state of hyperexcitability and play a critical role during FS (Ref. Reference Radzicki156). Elevated temperatures may affect most cellular events, including plasma membrane states and synaptic transmission (Refs Reference Thompson, Masukawa and Prince157, Reference Volgushev158). SV recycling has been reported to be temperature-associated. The SV size doubled and both endocytosis and exocytosis sped up at physiological temperatures higher than room temperature (Ref. Reference Micheva and Smith159).
FS usually occur in the condition of high or rapid rising body temperature in susceptible individuals. Genes associated with fever generation may be the possible causative factors for FS (Ref. Reference Saghazadeh160). Inflammatory cells and molecules, which are intrinsic to fever response, contribute to neuronal hyperexcitability and are involved in generating FS (Refs Reference Choy161, Reference Heida and Pittman162, Reference Dube163). Endogenous interleukin (IL)-1β is generated in the hippocampus during fever and promotes neuronal hyperexcitability via several mechanisms, which contribute to induction of seizure (Refs Reference Dube163, Reference Viviani164, Reference Vezzani165, Reference Vezzani166). Patients with IL-1β variants at position − 511 or in exon 5 are reported to be more susceptible to develop FS (Ref. Reference Virta, Hurme and Helminen167). Administration of high dose of IL-1β may induce seizures in immature wild-type mice. IL-1R1−/− mice have higher seizure threshold under hyperthermia environment (Refs Reference Dube163, Reference Feng168). Mice lacking IL-1β receptor type 1 are significantly resistant to FS generation (Ref. Reference Dube163). The variants of IL-1α−889 1/1, IL-1β−511 T/T, and IL6 SNPs increase the risk of FS (Refs Reference Saghazadeh160, Reference Shahrokhi169). IL-6 is the strong autoantibody production inducer, including IgA, IgM and IgG, which may occasionally play a role in the epileptogenesis (Refs Reference Vezzani166, Reference Vezzani170). Tumour necrosis factor-α may also be generated in the developing brain during FS and increase neuronal excitability (Ref. Reference Choy161). The specific mechanism of fever in provoking FS still remains widely debated.
Conclusions and future perspectives
The discovery of the disease-associated genes encoding the sodium channel subunits, such as SCN1A, SCN1B and SCN9A, and the genes encoding GABAA receptor subunits, such as GABRG2, GABRD and GABRB3, suggests that genetic factors are pivotal in FASE. Ion channels and neurotransmitter receptors are reasonable candidates for the seizure-causing alleles. However, not all families with FASE associate with these gene mutations. No disease-causing genes are identified in most families with FASE. Exclusion of ion channel genes and GABAA receptor subunit genes in FASE disease gene loci suggested that other presently unknown factors, beyond ion channel and neurotransmitter receptor abnormalities, are involved in the FASE molecular biology mechanisms. The incomplete penetrance and heterogeneous expression in FASE increase the difficulty of identifying disease genes. Recently, mutations in the several genes encoded synaptic proteins may be implicated in causing FASE, including the PRRT2 and STX1B genes. Normal synaptic transmission depends on a combination of the presynaptic neurotransmitter release machinery and corresponding postsynaptic receptors. The neurotransmitter release machinery involves in various functional proteins that mediate the SV formation, exocytosis and endocytosis recycling, which may take part in the generating of FASE. Elevated temperature may enhance synaptic transmission by promoting SV recycling and enlarging SV size. The inflammatory cells and molecules, which are intrinsic to the fever response, may contribute to neuronal hyperexcitability and be involved in triggering FS. These findings broaden the understanding of mechanism in FASE.
Mutations in certain genes may cause an overlapping spectrum of phenotypes ranging from isolated FS, FS+ and GEFSP to DS. According to the definition of International League Against Epilepsy (ILAE), FS are not proposed as an epileptic disorder for benign experiences and spontaneous resolutions prior to age 5. It is difficult to identify the prognosis of a single occurrence of FS based on short clinical characteristics. Several reported FEB families have been re-diagnosed with GEFSP via tracking the disease process and identifying of disease-causing genes. FEB3A, FEB3B and FEB8, caused by SCN1A, SCN9A and GABRG2 mutations, respectively, may best be classified as GEFSP. In view of the shared genetic foundations and the partial overlaps in phenotypes, FEB may be a mild GEFSP form.
Developing of next sequencing techniques, including genome-wide association studies, whole genome sequencing, exome sequencing and comparative genomic hybridisation chip analysis, will facilitate the discovery of new disease-causing genes or subtle gene variations of FASE. Combining data from genetic variants, gene expression, protein interaction and genetic animal models should shed new light on FASE mechanisms. This, in turn, may lead to developing new diagnostic and individual medical approaches.
Review criteria
References for this review were identified by searches of PubMed between 1977 and November 2017, and references from relevant articles. The search terms ‘febrile seizure’, ‘GEFSP’, ‘genetics’ and ‘epilepsy’ were used. There were no language restrictions. The final reference list was generated on the basis of relevance to the topics covered in this review.
Acknowledgements
This study was supported by grant 2016YFC1306604 from the National Key Research and Development Program of China; grant 81271921 and 81671296 from the National Natural Science Foundation of China; grants 2015JJ4088 and 2016JJ2166 from the Natural Science Foundation of Hunan Province, China; grant 20150301 from the New Xiangya Talent Project of the Third Xiangya Hospital of Central South University; and grant from the Foster Key Subject of the Third Xiangya Hospital of Central South University (Clinical Laboratory Diagnostics).
Competing interests
The authors declare no competing interests.