



Introduction
Essential tremor (ET) is a common movement disorder with reported prevalence of approximately 1% in the worldwide population (Ref. Reference Haubenberger and Hallett1). The age at onset appears to have a bimodal distribution with peaks around 2nd decade and 6th decade of life (Ref. Reference Kuhlenbäumer, Hopfner and Deuschl2). The original consensus statement of the Movement Disorder Society on tremor described the inclusion criteria of classic ET as follows: (i) bilateral, mostly symmetric postural or kinetic tremor, involving in hands and forearms and (ii) additional or isolated tremor involving in head, without abnormal posturing (Ref. Reference Deuschl3). Although there is no agreement among experts how to define ET, recent Movement Disorders Society ‘consensus’ statement proposed that ET is a syndrome with the following diagnostic criteria: (i) isolated tremor syndrome of bilateral upper limb action tremor; (ii) at least 3 years' duration; (iii) with or without tremor in other locations (e.g. head, voice or lower limbs) and (iv) the absence of other neurological signs, such as dystonia, ataxia or parkinsonism (Ref. Reference Bhatia4). Some patients start with ET syndrome but may later evolve into another tremor syndrome, such as Parkinson's disease (PD) (Ref. Reference Bhatia4). Such patients with ET–PD combinations have been classified as patients with PD and ‘antecedent ET’, but the relationship between the two disorders is not fully understood (Ref. Reference Tarakad and Jankovic5).
Another controversy in the field of ET is whether it is a chiefly physiological disorder resulting in abnormal function of the basal ganglia, thalamus, cerebellum and other structures or whether it is a neurodegenerative disorder. There is a growing support for the notion that the pathogenesis of ET is partly related to loss of Purkinje cells (PCs) and other pathological cerebellar abnormalities along with reduced γ-aminobutyric acid (GABA) receptor expression in the dentate nucleus (Ref. Reference Yuan6). Additionally, neurodegenerative changes such as Lewy bodies (LBs) reported in brains of some ET patients suggest the overlap between ET and PD (Refs Reference Tarakad and Jankovic5–Reference Louis7).
Although family history was thought not to be consistent enough to be included in the ET definition by the consensus statement (Ref. Reference Bhatia4), it is well recognised that genetic factors play a major role in ET pathogenesis. ET is familial in about 50–70% of cases (Ref. Reference Sullivan, Hauser and Zesiewicz8). First-degree relatives of ET patients were reported to have 4.7-fold risk of developing ET compared with controls (Ref. Reference Louis9). In twin studies, pairwise concordance was approximately two times in monozygotic twins, compared with dizygotic twins (Ref. Reference Tanner10). It has been proposed that ET is inherited as a complex disorder requiring interactions of genetic and non-genetic factors, but alternative explanations including polygenic inheritance and mitochondrial origin cannot be excluded (Ref. Reference Deng, Le and Jankovic11). At least 14 loci and 11 disease-causing genes related to ET have been described in the literature and reported in the Online Mendelian Inheritance in Man (OMIM, http://omim.org/) (Refs Reference Yuan6, Reference Leng12, Reference Liu13) (Table 1 and Fig. 1). In addition, many more genetic variants have been reported as risk or protective factors of ET, though the findings have been inconsistent (Refs Reference Yuan6, Reference Jiménez-Jiménez14, Reference Müller15) (Fig. 1).
Table 1. Summary of gene loci and disease-causing genes associated with essential tremor

AD, autosomal dominant; ET, essential tremor; PD, Parkinson's disease; SNP, single-nucleotide polymorphism, WES, whole exome sequence.
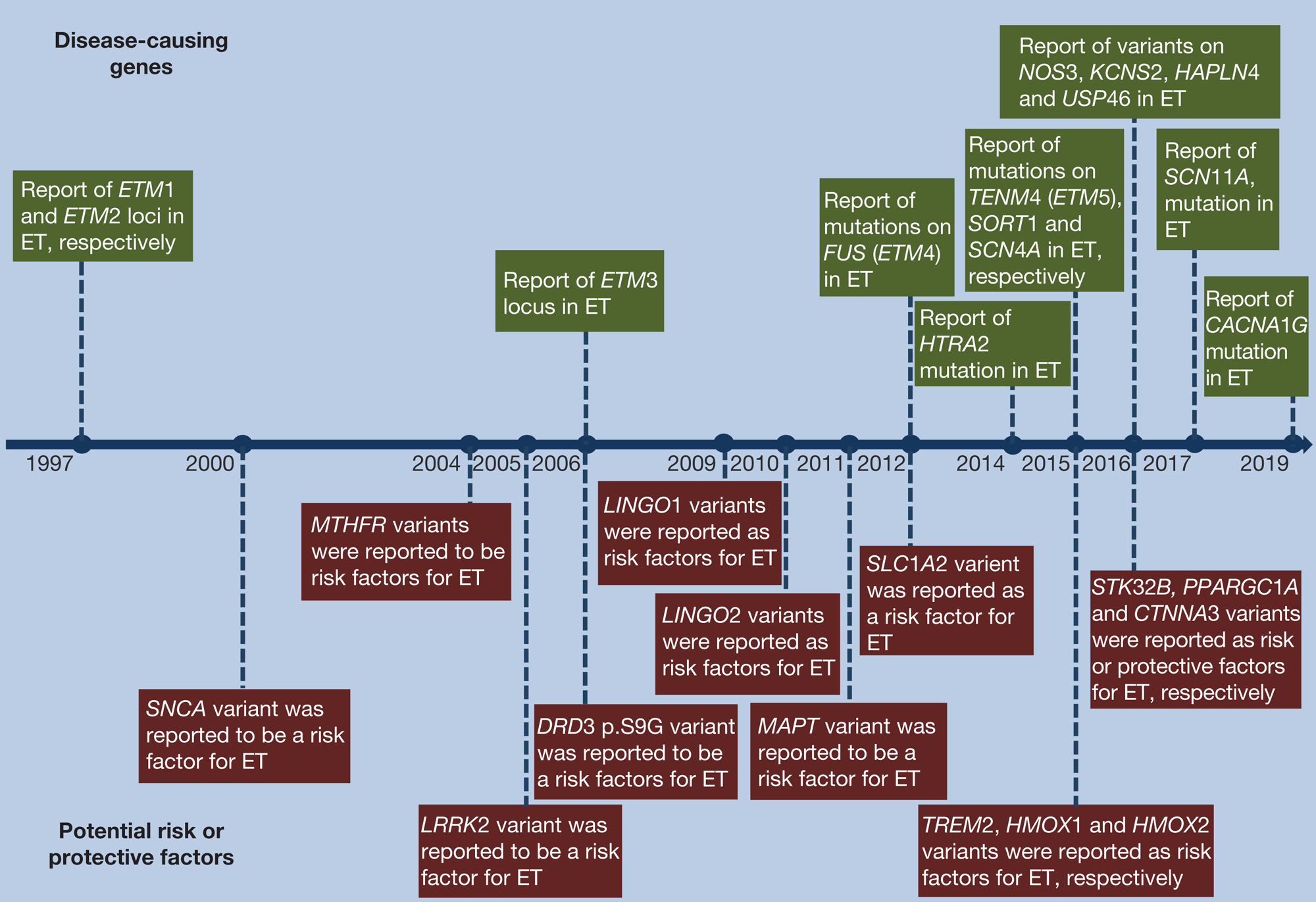
Fig. 1. Milestones in essential tremor research of disease-causing genes and potential risk or protective factors. The horizontal arrow in the middle of the figure represents the timeline. Disease-causing genes are shown above the timeline and potential risk or protective factors are shown below the timeline.
Although no ET-specific gene mutations have yet been identified, there is great interest in genetic testing in families with ET. In one study, 90/105 (85.7%) of families with ET expressed interest in being tested for possible genetic abnormalities (Ref. Reference Naranjo16). In this review we provide insights into possible genetic mechanisms of this common neurological disorder (Fig. 2), which, we hope, will lead to better diagnosis and novel treatment discoveries in future research of ET.

Fig. 2. An illustration of subcellular location and proposed function of gene products associated with essential tremor. FUS protein usually locates in nucleus and regulates nucleus–cytoplasm transport. TENM4 product, teneurin-4, was reported to enable its localisation to plasma membrane, and involved in neural development. HTRA2 protein, found in mitochondrion intermembrane space, promotes or induces cell death. Sortilin (SORT1 product), a protein usually regulating lipid metabolism, was found in plasma membrane and promotes neuronal apoptosis. Products of SCN4A and SCN11A mediate the voltage-dependent sodium ion permeability and membrane excitability, and products of KCNS2 can form functional heterotetrameric channels with KCNB1 and KCNB2, modulating the voltage-gated potassium channel activation then synaptic transmission. CACNA1G affects the calcium channel and ‘functional’ variants were recently found in ET families. NO synthase 3 (NOS3 product), highly expressed in cytoskeleton, plasma membrane and Golgi apparatus, was reported to participate in neuronal survival and synaptic plasticity. USP46 protein probably located in cytosol and BRAL2 protein (HAPLN4 product) located in extracellular matrix, regulate GABA action. GABA, γ-aminobutyric acid.
Genetic loci and gene analysis for ET
We define ‘variant’ is a neutral term including ‘mutation’ and ‘polymorphism’. The term ‘nucleotide mutation’, usually used in monogenic disorders, describes disease-causing or apparently disease-associated sequence variation. The term ‘polymorphism’, usually used in polygenic diseases, describes non-disease-causing sequence variation that has a frequency of at least 1% in the population (Ref. Reference Deng, Le and Jankovic11). We use ‘variant’ when it is not certain whether the sequence variation is a highly penetrant risk or is a confirmed risk factor for the disease.
Essential tremor 1 (ETM1, OMIM 190300)
ETM1, located on chromosome 3q13, was first found to be linked with ET by genome-wide scan of 16 Icelandic families in 1997 (Ref. Reference Gulcher17), and subsequently supported by findings in the Tajik families (Refs Reference Testa18, Reference Illarioshkin19). A p.S9G variant in the dopamine receptor D3 gene (DRD3) located in ETM1 was reported to confer inheritable susceptibility to ET in 23 French families (Ref. Reference Jeanneteau20) and in early-onset Caucasian patients from Spain (Ref. Reference García-Martín21). However, follow-up studies in Asian, Latvian, Italian, German, Danish, French and other populations with ET were not able to confirm this genetic susceptibility (Refs Reference Tan22–Reference Aridon30). Therefore, based on these irreproducible results, the DRD3 gene may, at best, be considered as a minor factor in ET susceptibility (Ref. Reference Vitale25).
A possible relationship has been found between dopamine D3 receptor (DRD3) and ET. DRD3, encoded by the DRD3 gene, usually highly expresses in the basal ganglia, and also expresses in the cerebellum, which has been implicated in the ET pathogenesis (Ref. Reference Jeanneteau20). In rat, DRD3 has been found to be expressed in the rat cerebellar PCs (Ref. Reference Diaz31). Several studies have provided support for the hypothesis that dysfunction in the cerebello–thalamo–cortical pathway played a role in the pathogenesis of ET, suggesting the expression level of DRD3 may be related to ET (Ref. Reference Lucotte32). DRD3 may be implicated in some neurologic movement disorders including PD and tardive dyskinesia (Refs Reference Lerer33, Reference Ryoo, Pierrotti and Joyce34), but further research is needed to better define the relationship between DRD3 and ET.
Essential tremor 2 (ETM2, OMIM 602134)
ETM2 was mapped to a 15-cM candidate region on chromosome 2p22-p25 in a Czech-American ET family in 1997 (Ref. Reference Higgins, Pho and Nee35). In a follow-up analysis adding three additional American families with ET, ETM2 was delimited to a 9.1-cM interval between the loci D2S224 and D2S405 by haplotype reconstruction (Ref. Reference Higgins36). In 2003, a haplotype on chromosome 2p24.1 formed by etm1231 and etm1234 was found to be segregated with ET (Ref. Reference Higgins37). Further analysis of ETM2 candidate region on chromosome 2p24.1 in Korean ET patients supports the linkage between ETM2 and ET (Ref. Reference Kim38). In 2005, the p.A265G variant (828C>G) in the HCLS1 binding protein 3 gene (HS1BP3), located in the minimal critical region (MCR) of ETM2, was reported as the causal variant in two unrelated ET families from America (Ref. Reference Higgins39). However, the p.A265G variant was not found to cosegregate with ET in a large Caucasian family in our study (Ref. Reference Deng40). Four recombination events were found in the <1 cM region between D2S2150 and etm1234, challenging the ETM2 MCR (Ref. Reference Deng, Le and Jankovic11). Reviewing previous studies, ETM2 may be mapped near the 9.1-cM interval between D2S224 and D2S405 (Refs Reference Higgins, Pho and Nee35–Reference Higgins41), and the p.A265G variant in the HS1BP3 gene is probably not responsible for monogenic ET (Ref. Reference Deng40).
Essential tremor 3 (ETM3, OMIM 611456)
In 2006, Shatunov et al. mapped the ETM3 locus to chromosome 6p23 by a combination of genome-wide linkage screening and fine mapping, but no pathogenic variant was identified after sequencing of 15 genes located in candidate regions (Ref. Reference Shatunov42). Given that the disease-associated haplotype was not present in some definite ET patients, and individuals harbouring the disease-associated haplotype didn't have fully developed ET in these families (Ref. Reference Shatunov42), a low penetrance or a high phenocopy rate should be considered in ET genetic expression if there is a monogenic causal gene in ETM3 (Ref. Reference Kuhlenbäumer, Hopfner and Deuschl2).
The FUS RNA binding protein gene (FUS, OMIM 137070) and essential tremor 4 (ETM4, OMIM 614782)
In 2012, p.Q290* mutation in the FUS gene on chromosome 16p11.2 was described to be responsible for ET by exome sequencing in a large ET-affected family (ETM4) (Refs Reference Merner43, Reference Mrózek, Karakousis and Bloomfield44). Another two variants, p.P431L and p.R216C, were identified in this gene by screening additional 270 ET patients (Ref. Reference Merner43). Further screening in our 180 Chinese Han patients detected a FUS p.M392I mutation (Ref. Reference Zheng45). Furthermore, a FUS p.R377W mutation was found by screening 217 Canadian cases (Ref. Reference Rajput46). However, the linkage between FUS and ET could not be replicated in other studies. No causal mutation was observed in 116 early-onset ET cases from America (Ref. Reference Parmalee47) or several other cohorts (Refs Reference Ortega-Cubero48, Reference Hedera49), and FUS might be a very rare cause of ET.
Although the exact function of the FUS protein is not clear, FUS appears to play a role in a series of cellular processes, including DNA repair, transcription regulation, cell proliferation, as well as RNA and microRNA processes (Ref. Reference Deng, Gao and Jankovic50). Even though FUS mutations may be a rare cause of ET, specific cellular and nervous system functions may be impaired by FUS mutations, possibly leading to loss of FUS function and development of ET (Refs Reference Merner43, Reference Schmouth, Dion and Rouleau51). FUS has two structural parts: nuclear localisation signal (NLS) and nuclear export signal (NES) sequences5, which play an important part in the nucleus–cytoplasm shuttle (Ref. Reference Schmouth, Dion and Rouleau51). The ET-related FUS mutation has been reported to be located in the NES motif, possibly interfering with FUS export from nucleus to cytoplasm via the nuclear pore complex (Ref. Reference Merner43). FUS mutations are also known to possibly cause other neurological disorders including amyotrophic lateral sclerosis (ALS) (Ref. Reference Macerollo and Bhatia52). The ET-causing mutations may be located in the NES and its transcripts are degraded by the nonsense-mediated-decay pathway (Ref. Reference Labbé53), whereas the ALS-causing mutations usually disrupt the NLS and lead to a truncated protein product (Refs Reference Macerollo and Bhatia52, Reference Labbé53).
Multiple animal models, including Drosophilae, zebrafish and mice, have provided powerful tools in studying the role of FUS loss-of-function in neurodegeneration. Drosophilae with human FUS-Q290* (hFUS-Q290*) may show specific characteristics that can be found in ET patients including age-related motor dysfunction, linked to the impairment of GABA-ergic pathway (Ref. Reference Tio54). In zebrafish, expression of mutant FUS or knock-down fus leads to locomotor impairment, and the fus-knock-down dysfunction can be rescued through co-expression of wild-type (WT) hFUS (Refs Reference Armstrong and Drapeau55, Reference Kabashi56). In mice, Fus−/− mice showed lymphocyte reduction and short survival time, but Fus+/− mice showed phenotypes undifferentiated from Fus+/+ animals (Ref. Reference Hicks57).
The teneurin transmembrane protein 4 gene (TENM4, OMIM 610084), essential tremor 5 (ETM5, OMIM 616736)
In 2015, disease-segregated mutations in the TENM4 gene mapped to chromosome 11q14.1 was identified in some Spanish ET families by whole exome sequence (WES) study (ETM5) (Ref. Reference Hor58). However, the previously described TENM4 p.A1442T mutation (Ref. Reference Hor58), was not found in any of 379 Chinese ET cases (but was found in two healthy individuals), suggesting that further studies of this variant are needed in larger ET cohorts and different populations (Ref. Reference Chao59).
TENM4, a member of the teneurin gene family, is highly expressed in the nervous system, and has been found to encode signalling molecule that functions as type-II transmembrane receptor at the cellular surface, or as a transcriptional regulator after intracellular domain export (Ref. Reference Tucker and Chiquet-Ehrismann60). TENM4 can regulate oligodendrocyte differentiation, and affect the small-diameter axon myelination in the central nervous system (CNS) (Ref. Reference Suzuki61). In mice, Tenm4 expression was found in the white matter of the cerebellum (Ref. Reference Zhou62). In vitro study with Oli-neu cells showed that the TENM4 mutations contribute to a TENM4 protein mislocalisation, from homogeneous membrane localisation to wrong-clustered localisation (Ref. Reference Hor58). This may hinder the TENM4-mediated phosphorylation and affect the oligodendroglial process outgrowth (Ref. Reference Hor58).
Synapse loss, as well as impairment in general synapse organisation and target select regulation was observed in Drosophilae with teneurin perturbations (Ref. Reference Mosca63). In transfected zebrafish, suppression and overexpression of TENM4 mRNA lead to defective branching and extension in the small diameter axon involving in truncal musculature (Ref. Reference Hor58). Tremors were only observed in Tenm4 −/− transgene mice but not in Tenm4 +/− mice (Ref. Reference Suzuki61). This suggests the possibility that the WT allele in heterozygous mice upregulates the Ten-4 expression and plays a compensating action (Ref. Reference Suzuki61). Tremor phenotype was presented in Tenm4 −/− mice, and its severity may reflect the dominant-negative effect of mutant TENM4 between haploinsufficiency and null phenotype (Refs Reference Hor58, Reference Suzuki61). In Tenm4 −/− mice, especially in the central nervous system (CNS) spinal cord, myelination was dramatically reduced in small-diameter axons, and oligodendrocyte differentiation was inhibited (Ref. Reference Suzuki61).
The HtrA serine peptidase 2 gene (HTRA2, OMIM 606441)
In 2014, a p.G399S mutation (c.1195G>A) in the HTRA2 gene was reported to be the causative mutation for ET in a six-generation Turkish family (Ref. Reference Unal Gulsuner64). Compared with heterozygotes, patients with homozygous HTRA2 p.G399S were linked to earlier age at onset and, severe postural/kinetic tremor with development of PD in the middle age (Ref. Reference Unal Gulsuner64). It should be noted that other groups, however, did not find the association between HTRA2 p.G399S and ET development in populations from Asian and Norway (Refs Reference He65–Reference Chao67).
The HTRA2 gene, located on chromosome 2p13 (Ref. Reference Gray68), codes for HTRA2, a serine protease located in mitochondrial intermembrane space (Ref. Reference Dagda and Chu69). It can be released into cytosol through an apoptotic stimulus and bind to the inhibitor of apoptosis proteins, initiating apoptosis (Refs Reference Unal Gulsuner64, Reference Dagda and Chu69). The HTRA2 p.G399S mutation was reported to change the mitochondrial morphology and function, reduce the protease activity and increase sensitivity to toxicity (Ref. Reference Strauss70). WT HtrA2 over-expressed mice showed a worse motor behavioural phenotype than HtrA2 p.G399S over-expressed mice, suggesting that HTRA2 p.G399S has a dominant-negative effect (Ref. Reference Casadei71).
The sodium voltage-gated channel alpha subunit 4 gene (SCN4A, OMIM 603967)
In 2015, a p.G1537S mutation in the SCN4A gene was found to be segregated with ET in a large Spanish family by WES study (Ref. Reference Bergareche72). SCN4A, mapped to chromosome 17q23.1-q25.3 (Ref. Reference Bennani-Baiti73), was generally considered as a muscle-specific gene, playing a major role in the pathogenesis of primarily myopathic disorders (Refs Reference Bergareche72, Reference Nicole and Fontaine74). It encodes the voltage-sensitive sodium channel (Nav1.4) protein that was also confirmed in the human cerebral cortex, suggesting the SCN4A expression in neuronal tissues (Ref. Reference Bergareche72). SCN4A p.G1537S mutation is situated in the IVS5-S6 loop portion, which inserts into the membrane and constructs the pore lining, playing an important role in ion selectivity of the sodium channel (Ref. Reference Bergareche72). Whole-cell patch-clamp studies showed a faster activation and a significantly faster near-threshold potential inactivation of channels with p.G1537S mutation, which could alter ion selectivity, reduce the repetitive action potential firing amplitudes, increase neural membrane excitability and accelerate the thalamic oscillations that manifest as tremor (Refs Reference Bergareche72, Reference Shaikh75). In addition, increased potassium and ammonium conductance was found in mutant protein, which may be involved in cortical inhibition impairment, causing neurological dysfunction and seizure activity (Ref. Reference Bergareche72).
The sortilin 1 gene (SORT1, OMIM 602458)
In 2015, a disease-segregating mutation p.G171A was identified in the SORT1 gene in a Spanish family with early-onset ET (Ref. Reference Sánchez76). SORT1, located on chromosome 1p13 (Ref. Reference Musunuru77), encodes sortilin, which is a member of the cellular vacuolar protein sorting 10 domain receptor family expressed in both CNS and peripheral nervous system (PNS) neurons (Ref. Reference Sánchez76). Sortilin acts as a neurotensin receptor, and plays a role in protein transport and signal transduction, regulating the viability and function of neurons (Refs Reference Nykjaer78, Reference Willnow, Petersen and Nykjaer79). It can also interact with the p75 neurotrophin receptor (p75NTR) and contribute to proNGF- and proBDNF-induced apoptosis of neurons (Ref. Reference Sánchez76). It has been reported that loss of sortilin activities may be associated with the development of several nervous system disorders including Alzheimer disease (Ref. Reference Willnow, Petersen and Nykjaer79). The SORT1 p.G171A mutation reduced both mRNA and protein level of SORT1 whereas the expression of its binding partner p75NTR was increased, which may cause abnormalities in neurotransmission deficiency and possibly tremor (Ref. Reference Sánchez76).
The sodium voltage-gated channel alpha subunit 11 gene (SCN11A, OMIM 604385)
In 2017, a p.R225C mutation in the SCN11A gene was identified in a four-generation Chinese family with early-onset episodic pain and adult-onset ET (Ref. Reference Leng12). SCN11A, located on chromosome 3p22.2, primarily expresses in the PNS nociceptors, and encodes Nav1.9 (Ref. Reference Leng12). The voltage-sensitive Na+ channels usually act as transmembrane proteins that play roles in action potential generation of excitable cells (Ref. Reference Ogata and Ohishi80). Nav1.9 regulates excitability of resting neurons by depolarising prolongation of subthreshold stimulus (Refs Reference Leng12, Reference Baker81, Reference Herzog, Cummins and Waxman82).
The nitric oxide synthase 3 gene (NOS3, OMIM 163729)
In 2016, variants p.G16S and p.P55L in the NOS3 gene were reported to be cosegregated with ET in two early-onset ET families, respectively (Ref. Reference Liu13). Further replicated results are needed to confirm the association between NOS3 and ET. NOS3, located on 7q36 (Ref. Reference Robinson83), encodes a major NO synthase isoform that highly expresses in the cerebellum, and plays a part in neurotransmitter NO conversion that affects NO-mediated neuronal survival and synaptic plasticity (Refs Reference Liu13, Reference de la Monte84). The NOS pathway has been reported to be involved in the development of several neurological disorders including PD and Alzheimer disease (Refs Reference de la Monte84, Reference Durrenberger85).
The potassium voltage-gated channel modifier subfamily S member 2 gene (KCNS2, OMIM 602906)
In 2016, a KCNS2 heterozygous p.D379E variant was reported to probably be the causal variant in an early-onset ET family by WES study, but subsequent Sanger sequencing on KCNS2 in additional 95 unrelated ET cases did not find any mutations (Ref. Reference Liu13). Follow-up replications are needed to confirm that association.
KCNS2 (KV9.2), encoded by the KCNS2 gene, is a K+ channel α subunit that is highly expressed in the cerebellar PCs and granular cells, and modulates both KV2.1 and KV2.2 channel activities (Refs Reference Liu13, Reference Salinas86). KCNS2 may regulate resting membrane potential and control the shape and frequency of action potentials (Ref. Reference Salinas86). This is supported by the similar function of Drosophila Shab, a highly homologous gene to KCNS2, regulating repetitive synaptic activities (Ref. Reference Ueda and Wu87). Mutations in Drosophila Shaker, which interacts with Drosophila Shab in synaptic transmission regulation, resulted in motor circuit abnormality, increased neuromuscular junction neurotransmission and leg shaking of Drosophilae under etherisation (Refs Reference Liu13, Reference Kaplan and Trout88), supporting the notion that K+ channel abnormities may be involved in ET pathology. Additionally, patients with mutant voltage-gated potassium channel gene (KCNA1, KV1.1), another member of K+ channel family, showed a tremor phenotype (Refs Reference Smith89, Reference Klein90).
The hyaluronan and proteoglycan link protein 4 gene (HAPLN4)
In one family with an early-onset ET, a p.G350R variant was reported as a probable causal variant in the HAPLN4 gene (Ref. Reference Liu13). Repetitive studies are required for confirmation. HAPLN4 is mainly expressed in GABA-ergic neurons including PCs and basket neurons in the cerebellar cortex (Ref. Reference Bekku91). Tremor has been linked to cerebellar neurodegeneration that was accompanied by PC loss, reduced activity of GABA system in deep cerebellar neurons, output disinhibition of deep cerebellar neurons that have pacemaker activity and the enhanced rhythmic activity of the thalamo-cortical circuit and thalamus (Ref. Reference Gironell92).
The ubiquitin specific peptidase 46 gene (USP46, OMIM 612849)
A heterozygous variant p.A133V in the USP46 gene was reported to be cosegregated with ET in an early-onset ET family, by WES study (Ref. Reference Liu13). Follow-up studies are needed for replication. USP46 is a deubiquitinating enzyme that is highly expressed in the cerebellum (Refs Reference Liu13, Reference Li93), which can regulate multilevel cellular functions by dissociating ubiquitin from protein substrates (Ref. Reference Zhang94). Impaired function of deubiquitinating enzyme family has been reported to lead to some neurodegenerative diseases such as PD (Ref. Reference Wu95), but not bipolar disorder and schizophrenia (Ref. Reference Kushima96). Mutant or knock-out Usp46 in mice have been found to have impaired regulation of GABA-ergic system, implicated in the pathophysiology of ET (Refs Reference Tomida97–Reference Helmich99).
The calcium voltage-gated channel subunit alpha1 G gene (CACNA1G, OMIM 604065)
Whole genome sequence and WES studies have recently identified ‘functional’ variants in the calcium voltage-gated channel subunit alpha1 G gene (CACNA1G, Cav3.1) in three ET families (Clark L., personal communication). Electrophysiologic studies by whole cell patch clamp recordings in HEK293T cells expressing the Cav3.1 mutant channels showed significant differences in the gating of the mutant Cav3.1 channels compared with the WT channel. Further studies are needed to confirm the sensitivity and specificity of this finding in populations of patients with ET, but T-type calcium channel modulators have been proposed to be effective in the treatment of ET (Ref. Reference Yang100).
Genetic risk or protective factors of ET
In addition to searching for monogenic causes of ET, genetic risk or protective factors also should be considered in the effort to better understand the pathogenesis of the disease.
The leucine rich repeat and Ig domain containing 1 gene (LINGO1, OMIM 609791)
In 2009, two ET-risk variants, rs9652490 and rs11856808, were reported in the LINGO1 gene by genome-wide association study (GWAS) of 452 Icelandic ET patients, and these variants were then found to have association with ET in follow-up samples from the United States, Germany, Austria and Iceland (Ref. Reference Stefansson101). Other subsequent studies, however, obtained incongruous results (Refs Reference Thier102–Reference Lorenzo-Betancor109). Intriguingly, whole-genome single-nucleotide polymorphism microarray analysis identified a genomic duplication encompassing the LINGO1 gene as the likely cause of familial dystonic tremor in a South Indian family recently (Ref. Reference Alakbarzade110), indicating that copy number variants (CNVs) may play an important role in the development of ET.
The LINGO1 gene, mapped to chromosome 15q24 (Ref. Reference Carim-Todd111), encodes a transmembrane glycoprotein specific to the CNS (Ref. Reference Deng, Gu and Jankovic112). It is highly expressed in neocortex, hippocampus, thalamus, amygdale and expressed in cerebellum and basal nuclei at lower levels (Ref. Reference Llorens113). The LINGO1 protein is involved in regulating neuronal survival, oligodendrocyte differentiation, axonal outgrowth and regeneration with a negative manner. Over-expression of LINGO1 has been found in the ET cerebellar cortex, and it appears to play a role in PC degeneration (Ref. Reference Agúndez114). CNS axonal myelination was found to develop earlier, and axonal integrity was improved in knock-out Lingo1 mice, with no obvious abnormalities of behaviour compared with WTs (Ref. Reference Mi115).
The leucine rich repeat and Ig domain containing 2 gene (LINGO2, OMIM 609793)
Variants rs1412229, rs7033345 and rs10812774 in the LINGO2 gene, a paralogue of LINGO1, were found to confer increased susceptibility for ET (Refs Reference Vilariño-Güell106, Reference Wu116).
In mice, the LINGO2 protein was found in the brain and seems to be restricted to neuronal tissue (Refs Reference Haines and Rigby117, Reference Homma118). Owing to the high homology, LINGO2 may present similar functions as LINGO1 and play an important role in maintenance of dopaminergic integrity (Ref. Reference Wu116).
The solute carrier family 1 member 2 gene (SLC1A2, OMIM 600300)
In 2012, variant rs3794087 in the SLC1A2 gene was reported to be associated with ET susceptibility by GWAS of 990 cases and 1537 controls from Europe (Ref. Reference Thier119). However, this variant appeared to be related to ET protection in a Chinese cohort (Ref. Reference Tan120), suggesting that linkage disequilibrium exists in different races, and rs3794087 is only a genetic marker reflecting SLC1A2 or its nearby gene associated with ET susceptibility.
The SLC1A2 gene, located on chromosome 11p13-p12 (Ref. Reference Li and Francke121), primarily encodes a glial high-affinity glutamate reuptake transporter called ‘excitatory amino acid transporter type 2’ (EAAT2) in the brain (Refs Reference Thier119, Reference Lee122). EAAT2 is expressed around the PC axon initial segment. In ET cases, EAAT2 is significantly reduced in cerebellar cortex (Ref. Reference Lee122). EAAT2 function is to protect glutamatergic olivo-cerebellar climbing fibres from excitotoxic injury, which can lead to PC excitotoxic death, postulated to be involved in ET pathogenesis (Refs Reference Schmouth, Dion and Rouleau51, Reference Lee122). Slc1a2 −/− mice showed high susceptibility of acute cortical injury in the brain, as well as epilepsy (Ref. Reference Tanaka123).
The serine/threonine kinase 32B gene (STK32B)
In 2016, GWAS in 2807 European patients and 6441 controls found that C allele of rs10937625 in the STK32B gene acts as a protective factor of ET (Ref. Reference Müller15). This positive result was replicated by analysis of 218 cases and 315 controls of Han Chinese population (Ref. Reference Zhang124).
The STK32B gene, located on chromosome 4p16 (Ref. Reference Ingersoll125), encodes the serine/threonine kinase. The STK32B expression, which was increased in ET cerebellar cortex, was found to be reduced with the rs10937625 variant, located on the DNase hypersensitive place of STK32B, supporting possible protective role of the rs10937625 C allele in development of ET (Refs Reference Müller15, Reference Zhang124).
The PPARG coactivator 1 alpha gene (PPARGC1A, OMIM 604517)
The rs17590046 variant in the PPARGC1A gene was found to be related to the susceptibility of ET by GWAS (Ref. Reference Müller15). However, conflicting results were described in two independent studies in Asian populations (Refs Reference Zhang124, Reference Xiao126).
The PPARGC1A gene, located on chromosome 4p15.1 (Ref. Reference Esterbauer127), encodes a transcriptional coactivator called the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), which plays a role in mitochondrial function and energy metabolism (Ref. Reference Müller15). PGC-1α can regulate oxidative metabolism and oxidative capacity-associated biological programmes, and loss of PGC-1α function may result in neuronal degeneration in the brain, especially in the striatum (Ref. Reference Lin128). Altered-function of PGC-1α has been reported to be associated with several neurodegenerative movement disorders including PD and Huntington's disease (HD), which share similar GABAA receptor abnormality as described in ET (Refs Reference Zheng129–Reference Boecker131). Motor impairment and striatal vacuolation are found in knocked-out PGC-1α mice, suggesting that PGC-1α plays an important role in motor function (Ref. Reference Lucas132).
The catenin alpha 3 gene (CTNNA3, OMIM 607667)
The rs12764057, rs10822974 and rs7903491 variants in the CTNNA3 gene were reported to be associated with ET susceptibility by GWAS in European patients (Ref. Reference Müller15). Subsequently, the G allele of the CTNNA3 rs7903491 variant was confirmed as an ET risk factor whereas the association of ET for rs12764057 and rs10822974 could not be duplicated in a Chinese cohort (Ref. Reference Zhang124).
CTNNA3, located on chromosome 10q21 (Ref. Reference Janssens133), encodes a cell–cell adhesion molecule named catenin alpha 3 (Ref. Reference Müller15), which is primarily expressed in testis and heart, with lower expression in the brain (Ref. Reference Janssens134). CTNNA3 has been reported to be involved in Alzheimer disease development (Ref. Reference Miyashita135), though the specific pathogenic pathway is not clear. There is some epidemiological evidence for possible clinical and pathological overlap between ET and Alzheimer disease, but further studies are needed to establish the link (Refs Reference Schmouth, Dion and Rouleau51, Reference LaRoia and Louis136).
Risk factors associated with other neurological diseases
ET phenotype has been reported to possibly overlap with other neurological diseases including PD, Alzheimer disease, HD, ALS, dystonia and myoclonus (Refs Reference Deng, Le and Jankovic11, Reference Schmouth, Dion and Rouleau51, Reference Macerollo and Bhatia52, Reference Boecker131). These reported associations have been challenged, but research into possible genetic causes of ET and related neurological diseases may provide new insights into the relationship between ET and other diseases.
Based on many epidemiologic and other studies, the support for ET–PD relationship is stronger than the other associations (Ref. Reference Benito-León, Louis and Bermejo-Pareja137). In addition, pathologic studies have found LB pathology in brains of ET patients (Ref. Reference Louis7), providing additional support for the relationship between ET and PD. A variety of variants on genes that are related to PD have been reported as risk factors for ET susceptibility, including the synuclein alpha gene (SNCA) (Ref. Reference Tan138), the leucine rich repeat kinase 2 gene (LRRK2) (Ref. Reference Skipper139), the methylenetetrahydrofolate reductase gene (MTHFR) (Ref. Reference Sazci, Ergul and Bayulkem140), the heme oxygenase 1 gene (HMOX1), the heme oxygenase 2 gene (HMOX2) (Ref. Reference Ayuso141), the microtubule associated protein tau gene (MAPT) (Ref. Reference Vilariño-Güell142) and the triggering receptor expressed on myeloid cells 2 gene (TREM2) (Ref. Reference Ortega-Cubero143).
ET-like tremor was also found in X-linked spinal and bulbar muscular atrophy, type 1 (SMAX1, also known as Kennedy disease), fragile X tremor/ataxia syndrome (FXTAS), spinocerebellar ataxia type 12 (SCA-12) and some other neurological diseases (Ref. Reference Deng, Le and Jankovic11). Variants in the related genes such as the androgen receptor gene (AR) (Ref. Reference La Spada144), the fragile X mental retardation 1 gene (FMR1) (Ref. Reference Jacquemont145), the protein phosphatase 2 regulatory subunit β gene (PPP2R2B, also known as SCA12), the peripheral myelin protein 22 gene (PMP22), the chromosome 9 open reading frame 72 gene (C9orf72), the ataxin 2 gene (ATXN2), and the ataxin 3 gene (ATXN3) may shed light on ET genetic risk factors (Ref. Reference Kuhlenbäumer, Hopfner and Deuschl2). However, abnormalities in these genes have not been consistently found in ET population studies and their pathogenic relationship to ET, if any, is doubtful.
Genetic factors suggested by animal models
Animal models provide important insights into the pathogenesis of ET and may contribute to the development of treatment. Reduced GABA levels in the cerebrospinal fluid of ET patients (Ref. Reference Málly, Baranyi and Vizi146), as well as the well-recognised tremor suppression by alcohol and other GABA-ergic drugs including benzodiazepines, suggests that the disturbed GABA-ergic pathway may play a role in ET pathogenesis (Refs Reference Mostile and Jankovic147, Reference Jankovic148). Postural/kinetic tremor and motor incoordination, which are characteristics of ET, were reported in GABAA receptor α1−/− (Gabra1−/−) mice (Refs Reference Kralic149, Reference Jankovic and Noebels150), but no susceptibility association has been found after genetic analysis of the gamma-aminobutyric acid type A receptor alpha1 subunit gene (GABRA1) in ET cases (Ref. Reference Deng151). In addition, motor impairments including gait and balance abnormalities and continuous tremor were reported in GABA transporter subtype 1 gene (Gat1) knock-out mice (Ref. Reference Chiu152). However, no ET risk factor had been found in GABA transporter or receptor genes (Ref. Reference Thier153).
Mutant rats with both the tremor (tm) mutation and the hyperpolarisation-activated cyclic nucleotide-gated 1 channel (Hcn1) mutation showed spontaneous tremors resembling ET, whereas rats carrying mutant tm or Hcn1 alone presented no tremor, indicating ET may be a digenic inherited disease. Blocking HCN1 channels resulted in kinetic tremor in tm mutant rats, confirming the important role of Hcn1 (Ref. Reference Ohno154). HCN1, a member of the HCN channel family expressed in heart and the nervous system (Ref. Reference Biel155), can be activated by membrane hyperpolarisation, indicating a role in transportation of sodium and potassium ions (Ref. Reference Ohno154). The mutant HCN1 leads to restraint of hyperpolarisation-activated currents, suggesting a loss-of-function mechanism (Ref. Reference Ohno154). Several mutations in the sodium voltage-gated channel alpha subunit 8 gene (Scn8a) were found to result in congenital postural and kinetic tremor in mice of the extremities, as well as ataxia and dystonia, but after SCN8A screening of 95 Caucasian patients with familial ET, no causal mutation had been detected (Refs Reference Meisler156, Reference Sharkey157).
The deficiency of a mouse PC gene, carbonic anhydrase 8 (Car8), was found to cause an ET-like tremor. The tremor mimics the ET condition of human in oscillation frequency, age-related progression and alcohol responsiveness (Refs Reference Kuo158, Reference White159). Mutations in human carbonic anhydrase 8 gene (CA8), a homologue of Car8, was reported to result in tremor in addition to ataxia (Refs Reference Kuo158, Reference Türkmen160), and the specific mechanisms need further studies.
Therapy
Therapy of ET is still symptomatic (Ref. Reference Hedera, Cibulčík and Davis161), but the goal of current research is to find a pathogenesis-targeted treatment in the future. Therapy of ET can be roughly grouped into categories of pharmacologic, surgical and other nonpharmacologic or nonsurgical treatment approaches (Ref. Reference Haubenberger and Hallett1). Pharmacologic therapy, including four first-line drugs, such as propranolol, primidone, gabapentin and topiramate, is considered when tremor impacts daily living or psychological health (Refs Reference Sullivan, Hauser and Zesiewicz8, Reference Lorenz and Deuschl162). Surgical therapy, such as thalamic deep brain stimulation or focused ultrasound thalamotomy, is reserved for drug-intolerant or drug-resistant patients with troublesome or disabling ET (Refs Reference Wharen163–Reference Thanvi, Lo and Robinson165).
Future strategy
Genetic testing for ET has not reached mainstream clinical practice though several variants have been found in patients and families with ET. The plausible reasons include inconsistent definition of ET, ET misdiagnosis, age-related occurrence, complex genetic basis, incomplete penetrance, epigenetic modification, environmental disturbance and other factors, which may make it difficult to identify specific ET-related genes by current strategies (Ref. Reference Tan and Schapira166). ET is likely a syndrome with different subtypes and aetiologies. Observation of four disease-associated channel gene mutations (SCN4A p.G1537S, SCN11A p.R225C, KSCN2 p.D379E and CACNA1G variants) in ET families, as well as mutant Hcn1, and Scn8a tremor animal models, indicates that different channel abnormalities may be responsible for subtypes of ET (Refs Reference Leng12, Reference Liu13, Reference Bergareche72, Reference Ohno154, Reference Meisler156).
ET genetic studies may provide insights into the pathogenesis of the common disorder, improve diagnosis and eventually lead to more specific pathogenesis-targeted therapies. Analyses based on large sample size, diversified populations, CNV examination, modern omics sequencing techniques and cellular/system-level studies should be performed to investigate potential pathogenic and pathophysiological mechanisms of ET (Refs Reference Kuhlenbäumer, Hopfner and Deuschl2, Reference Clark and Louis167).
Author contributions
H.D., S.W. and J.J. contributed equally to researching data for the paper, discussions of the content and writing the article. H.D. and J.J. contributed to reviewing and editing of the manuscript before submission.
Financial support
H.D.'s research was supported by grants from the National Key Research and Development Program of China (2016YFC1306604), the National Natural Science Foundation of China (81873686), the Science and Technology Program of Hunan Province (2017SK50131). S.W.'s research was supported by Research Innovation Program for Graduate Students of Central South University (2019zzts831). J.J. receives support from the Parkinson Foundation and Michael J. Fox Foundation for Parkinson Research.
Conflict of interest
The authors declare no conflict of interest.



