


Schizophrenia does not typically emerge until adolescence, but the etiology of the disorder may begin before birth. This link was first indicated by the identification of maternal influenza during pregnancy as a risk factor for schizophrenia in epidemiologic studies (Brown, Begg, et al., Reference Brown, Begg, Gravenstein, Schaefer, Wyatt, Bresnahan and Susser2004; Mednick, Machon, Huttunen, & Bonett, Reference Mednick, Machon, Huttunen and Bonett1988). Soon after, infections with other pathogens were identified as risk factors, including rubella (Brown et al., Reference Brown, Cohen, Harkavy-Friedman, Babulas, Malaspina, Gorman and Susser2001), herpes simplex virus (Buka, Cannon, Torrey, & Yolken, Reference Buka, Cannon, Torrey and Yolken2008; Mortensen et al., Reference Mortensen, Nørgaard-Pedersen, Waltoft, Sørensen, Hougaard, Torrey and Yolken2007), and toxoplasmosis (Brown et al., Reference Brown, Schaefer, Quesenberry, Liu, Babulas and Susser2005). The fact that a wide variety of infections can contribute to the development of the same disorder suggests that maternal immune activation, and subsequent inflammation in the fetus, may mediate the association between prenatal infection and risk for schizophrenia (Meyer, Reference Meyer2013). Inflammatory responses in the fetal brain can be induced by maternal bacterial (Cai, Pan, Pang, Evans, & Rhodes, Reference Cai, Pan, Pang, Evans and Rhodes2000) and viral (Stridh et al., Reference Stridh, Mottahedin, Johansson, Valdez, Northington, Wang and Mallard2013) infections, as well as birth complications such as hypoxia/ischemia (Girard, Larouche, et al., Reference Girard, Larouche, Kadhim, Rola-Pleszczynski, Gobeil and Sébire2008). Recent evidence suggests that inflammation may also mediate the relationship with other prenatal risk factors for schizophrenia (Miller, Culpepper, Rapaport, & Buckley, Reference Miller, Culpepper, Rapaport and Buckley2013), including malnutrition (Brown & Susser, Reference Brown and Susser2008) and maternal stress (van Os & Selten, Reference van Os and Selten1998). It is important to point out that the vast majority of individuals exposed to maternal immune activation do not develop schizophrenia (Cannon et al., Reference Cannon, van Erp, Rosso, Huttunen, Lönnqvist, Pirkola and Standertskjöld-Nordenstam2002). This suggests that exposure to prenatal inflammation interacts with other risk factors in vulnerable individuals to increase risk for the disorder. Variants in immune genes are strongly implicated in genetic studies of risk for schizophrenia (Arion, Unger, Lewis, Levitt, & Mirnics, Reference Arion, Unger, Lewis, Levitt and Mirnics2007; Ripke et al., Reference Ripke, Neale, Corvin, Walters, Farh, Holmans and O'Donovan2014). However, genetic risk for schizophrenia does not seem to be associated with increased propensity for prenatal complications associated with inflammation, including maternal infection or stress during pregnancy and hypoxia-associated obstetric complications (Mittal, Ellman, & Cannon, Reference Mittal, Ellman and Cannon2008). It is possible that exposure to maternal immune activation increases risk for schizophrenia by interacting with underlying genetic vulnerability to immune disruption (Clarke, Tanskanen, Huttunen, Whittaker, & Cannon, Reference Clarke, Tanskanen, Huttunen, Whittaker and Cannon2009).
Broadly, prenatal inflammatory exposure may influence neurodevelopment by affecting immune cell functioning in the brain. Peripheral inflammation activates microglia, immune cells in the central nervous system (CNS) that secrete inflammatory proteins that help resolve inflammation but can be toxic in large amounts (Deverman & Patterson, Reference Deverman and Patterson2009; Ratnayake, Quinn, Walker, & Dickinson, Reference Ratnayake, Quinn, Walker and Dickinson2013). Active microglia are involved in the development of cellular mechanisms implicated in schizophrenia, such as synaptic connections between pyramidal neurons (Paolicelli et al., Reference Paolicelli, Bolasco, Pagani, Maggi, Scianni, Panzanelli and Gross2011), and overactivation of microglia could damage these systems (Monji et al., Reference Monji, Kato, Mizoguchi, Horikawa, Seki, Kasai and Kanba2013). Disruption to synapses and other cellular processes are thought to impair cognitive function and could contribute to the psychological symptoms of schizophrenia (Feinberg, Reference Feinberg1982; Weinberger, Reference Weinberger1987). This suggests that activation of microglia during early development could set individuals on a neurodevelopmental risk trajectory that ultimately leads to the development of the disorder (Insel, Reference Insel2010). In addition, prenatal inflammatory events are thought to “program” microglia to be more reactive to future immune challenges, which may cause compounding damage as neural systems and circuits mature and are refined throughout later childhood and adolescence (Monji, Kato, & Kanba, Reference Monji, Kato and Kanba2009; Patterson, Reference Patterson2009). Exaggerated inflammatory responses to insults in adolescence, such as stress or infection, could cause additional damage to vulnerable neural systems and contribute to the proximal onset of schizophrenia (Bergink, Gibney, & Drexhage, Reference Bergink, Gibney and Drexhage2014). This theory, termed the “multiple hits” theory, has gained support from rodent and human studies of risk for schizophrenia (for review see Bayer, Falkai, & Maier, Reference Bayer, Falkai and Maier1999; Bilbo & Schwarz, Reference Bilbo and Schwarz2009; Meyer, Reference Meyer2013).
Converging evidence suggests a role for inflammation in the etiology of schizophrenia. However, the specific inflammatory pathways that mediate this association remain unclear. Potential candidate systems include inflammatory proteins produced by microglia, such as cytokines. Cytokines are involved in initiating and regulating inflammatory responses in the periphery and the brain, can be toxic to neurons in large amounts, and are implicated in neurodegenerative disorders such as Alzheimer's (Deverman & Patterson, Reference Deverman and Patterson2009; Stephan et al., Reference Stephan, Barres and Stevens2012). In addition, low concentrations of cytokines secreted by microglia have been implicated in neurodevelopmental processes such as synaptic formation and elimination (Gilmore, Fredrik Jarskog, Vadlamudi, & Lauder, Reference Gilmore, Fredrik Jarskog, Vadlamudi and Lauder2004). Specific cytokines may be implicated in typical neurodevelopmental processes (Deverman & Patterson, Reference Deverman and Patterson2009), such as IL-6 in the development of small dendrite projections on cortical neurons that provide the basis for synaptic formation (Gilmore et al., Reference Gilmore, Fredrik Jarskog, Vadlamudi and Lauder2004). Rodent models suggest that prenatal cytokine exposure can also have lasting influence on cognitive function. For example, elevated IL-6 during mid-pregnancy can cause deficits in prepulse and latent inhibition in adult offspring (Smith, Li, Garbett, Mirnics, & Patterson, Reference Smith, Li, Garbett, Mirnics and Patterson2007) and elevated IL-10 at a similar point during pregnancy can impair spatial exploration and associative learning in adulthood (Meyer et al., Reference Meyer, Murray, Urwyler, Yee, Schedlowski and Feldon2008). Thus, cytokines are a strong candidate mechanism for linking prenatal inflammation with the etiology of schizophrenia.
Recently, a second candidate mechanism has emerged as well. The complement system is a component of the innate immune system that produces proteins involved in “tagging” and deactivating pathogens and damaged cells for removal by other immune cells (Mayilyan, Weinberger, & Sim, Reference Mayilyan, Weinberger and Sim2008; Stephan, Barres, & Stevens, Reference Stephan, Barres and Stevens2012). Microglia and astrocytes produce large quantities of complement proteins in the CNS in response to indicators of injury or inflammation (Stephan et al., Reference Stephan, Barres and Stevens2012). Neurons can be damaged by the complement system (Orsini, de Blasio, Zangari, Zanier, & de Simoni, Reference Orsini, de Blasio, Zangari, Zanier and de Simoni2014), and uncontrolled complement activation has been observed in neurodegenerative disorders (Orsini et al., Reference Orsini, de Blasio, Zangari, Zanier and de Simoni2014). Emerging evidence suggests that complement proteins may also be implicated in neurodevelopmental processes, including neuronal differentiation and migration and synaptic pruning (Mayilyan et al., Reference Mayilyan, Weinberger and Sim2008; Sekar et al., Reference Sekar, Bialas, de Rivera, Davis, Hammond, Kamitaki and McCarroll2016; Stephan et al., Reference Stephan, Barres and Stevens2012). Aside from one study linking elevations in complement protein C1q at birth to risk for schizophrenia (Severance, Gressitt, Buka, Cannon, & Yolken, Reference Severance, Gressitt, Buka, Cannon and Yolken2014), the long-term consequences of prenatal complement exposure on neurodevelopment in humans are relatively unexplored. The similarities between the cytokine and complement systems suggest that complement may also be a mechanism linking prenatal inflammation with neurodevelopmental changes implicated in schizophrenia.
Further investigation of links between prenatal alterations of cytokine and complement pathway activity and risk for schizophrenia may yield additional specificity into neurodevelopmental pathways that are implicated in the etiology of schizophrenia. As different components of the brain develop during distinct periods of prenatal development, it is possible that elevations in cytokines and complement proteins at different time points could have different effects on neurodevelopment (Meyer, Yee, & Feldon, Reference Meyer, Yee and Feldon2007). Ultimately, these different prenatal insults may lead to disruption of a similar set of cognitive processes and the emergence of the disorder (i.e., equifinality; Cicchetti & Rogosch, Reference Cicchetti and Rogosch1996). Understanding the distinct pathways that ultimately lead to the symptoms of schizophrenia could yield more targeted and specific interventions that could help prevent the development of schizophrenia in individuals at high risk.
This review focuses on two candidate mediators of the relationship between microglial activation in response to maternal immune activation and risk for schizophrenia: cytokines and complement. We consider how their roles in typical neurodevelopment might help clarify risk pathways that mediate the association between prenatal immune insults and schizophrenia. We start by reviewing the cellular and structural abnormalities associated with the development of schizophrenia and discuss how these abnormalities may give rise to the psychological symptoms of the disorder. We then discuss prenatal neurodevelopment and how the continued refinement of neural organization gives rise to increasingly complex cognition in childhood and adolescence. We review the literature on prenatal cytokine and complement exposure and risk for schizophrenia, and identify specific proteins and time points that are implicated in risk. We then discuss the known roles of cytokines and complement proteins in these normative processes, and discuss how alterations in these pathways due to immune activation at distinct time points during prenatal development may increase risk for schizophrenia. We use this information to consider how these immune insults could affect childhood and adolescent developmental risk trajectories for psychosis, and identify areas of further research needed to support neonatal interventions to encourage healthy neurodevelopment.
Characterization of Schizophrenia
Presumably, inflammation increases risk for schizophrenia by influencing aspects of neurodevelopment that play a role in the disorder. To assess the viability of this notion, it is helpful to consider what is currently known about schizophrenia. Clinically, schizophrenia is characterized by positive symptoms, such as delusions and hallucinations, and negative symptoms, such as lack of motivation and anhedonia. Cognitive deficits, such as poor working memory and processing speed, are also associated with the disorder (American Psychiatric Association, 2013). The development of these symptoms is associated with deficits in a variety of cognitive processes observed in these individuals, including memory, language, executive function, attention, and intelligence (Fioravanti, Carlone, Vitale, Cinti, & Clare, Reference Fioravanti, Carlone, Vitale, Cinti and Clare2005). These deficits are thought to reflect a failure to integrate information from functionally distinct regions of the brain, which may emerge from a general disconnectivity between neurons (Friston, Brown, Siemerkus, & Stephan, Reference Friston, Brown, Siemerkus and Stephan2016; Friston & Frith, Reference Friston and Frith1995). This disconnectivity is thought to be associated with abnormalities in neural networks, which may reflect deficits in neural structure and function within and between regions (Friston et al., Reference Friston, Brown, Siemerkus and Stephan2016; Friston & Frith, Reference Friston and Frith1995). Neuroimaging studies of functional connectivity suggest that schizophrenia is broadly characterized by weaker and more diverse functional connections between regions compared to healthy individuals, and that these alterations in functional activity may contribute to poorer cognition (Lynall et al., Reference Lynall, Bassett, Kerwin, McKenna, Kitzbichler, Muller and Bullmore2010). Similarly, many different structural changes have been observed in schizophrenia, including reduction in gray matter volume in regions such as the medial temporal lobe, much of the cortex, basal ganglia, and thalamus; decreased white matter volume and tract coherence across the brain; and expanded ventricles (Davis, Stewart, Friedman, & Buchsbaum, Reference Davis, Stewart, Friedman and Buchsbaum2003; Shenton, Dickey, Frumin, & McCarley, Reference Shenton, Dickey, Frumin and McCarley2001). Emerging evidence suggests that functional and structural neural deficits may reflect abnormalities in cellular structure and activity of neurons, including synaptic density and number and type of synaptic connections between cortical neurons (Reimann et al., Reference Reimann, Nolte, Scolamiero, Turner, Perin, Chindemi and Markram2017; van den Heuvel, Scholtens, de Reus, & Kahn, Reference van den Heuvel, Scholtens, de Reus and Kahn2016). As inflammatory pathways are thought to affect these cellular processes that ultimately give rise to the neural, cognitive, and behavioral changes observed in schizophrenia, we will focus further on cell-level deficits in development of schizophrenia.
Neurobiological deficits associated with schizophrenia onset
To understand the etiology of schizophrenia, it is important to identify which neurological changes are mechanistically related to the emergence of the disorder and which changes may be only correlated or are secondary to medication effects (Cannon, Reference Cannon2015). One approach to identifying potential mechanisms is to prospectively study adolescents at clinical high risk for schizophrenia to identify changes that correlate with the onset of the disorder. Studies with this design have identified reductions in gray matter volume, particularly in the prefrontal cortex, temporal cortex, and hippocampus (Cannon et al., Reference Cannon, Chung, He, Sun, Jacobson, van Erp and Heinssen2015; Satterthwaite et al., Reference Satterthwaite, Wolf, Calkins, Vandekar, Erus, Ruparel and Gur2016), that occur proximally to the onset of psychosis. These regions are implicated in a wide variety of cognitive processes, including selective attention, behavioral inhibition, working memory, long-term declarative memory, sensory integration, and social cognition (Hein & Knight, Reference Hein and Knight2008; Miller & Cohen, Reference Miller and Cohen2001; Squire, Reference Squire1992). In addition, reduced gray matter volumes in these regions have been associated with cognitive deficits in individuals with schizophrenia, such as poorer performance on word memory tasks among patients with smaller hippocampal volumes (Sanfilipo et al., Reference Sanfilipo, Lafargue, Rusinek, Arena, Loneragan, Lautin and Wolkin2002). Postmortem studies of individuals with schizophrenia suggest that the observed reductions in gray matter are driven by decreased synaptic density and interneuron cell populations (Bennett, Reference Bennett2011), and perhaps also by increases in white matter volume (Catts et al., Reference Catts, Fung, Long, Joshi, Vercammen, Allen and Shannon Weickert2013). Examining ways in which these cellular mechanisms may contribute to schizophrenia can provide a platform for investigating mechanisms by which inflammation may contribute to the development of the disorder.
Synaptic density
One component of gray matter is synapses. Synapses are connections between neurons that are formed on protruding dendritic spines and facilitate communication (i.e., excitation or inhibition of the downstream neuron) that ultimately give rise to cognitive function (Glausier & Lewis, Reference Glausier and Lewis2013). Reductions in excitatory glutamatergic synapses and dendritic spine density have been consistently observed in the dorsolateral prefrontal cortex of individuals with chronic schizophrenia, and may also be reduced in the medial prefrontal cortex, superior temporal cortex, and hippocampus (Broadbelt, Byne, & Jones, Reference Broadbelt, Byne and Jones2002; Garey et al., Reference Garey, Ong, Patel, Kanani, Davis, Mortimer and Hirsch1998; Harrison & Eastwood, Reference Harrison and Eastwood2001). In the prefrontal cortex, these deficits are most consistently observed in excitatory pyramidal neurons in layers III and possibly layer V of the dorsolateral and medial prefrontal cortex. These layers are thought to contain synapses connecting cortical pyramidal neurons and GABAergic interneurons as well as synapses connecting cortical and thalamic neurons (Broadbelt et al., Reference Broadbelt, Byne and Jones2002; Glantz & Lewis, Reference Glantz and Lewis2000). Reduced synaptic connectivity in these layers and regions may be associated with functional and cognitive deficits observed in schizophrenia. For example, reductions in layer III pyramidal spine density in cortical regions including the superior frontal cortex, orbitofrontal cortex, and portions of the temporal cortex may be associated with poorer cortical white matter connectivity in individuals with schizophrenia (van den Heuvel et al., Reference van den Heuvel, Scholtens, de Reus and Kahn2016). Broadly, synaptic connections in layer III of the dorsolateral prefrontal cortex form microcircuits of cortical pyramidal neurons that provide the backbone of working memory (Arnsten, Reference Arnsten2011). Though specific cellular mechanisms of other cognitive processes are less clearly determined or tied to specific regions or layers, activity within the medial prefrontal cortex and superior temporal cortex has been associated with cognitive processes such as memory retrieval and source monitoring (Brandt, Bergström, Buda, Henson, & Simons, Reference Brandt, Bergström, Buda, Henson and Simons2014; Sugimori, Mitchell, Raye, Greene, & Johnson, Reference Sugimori, Mitchell, Raye, Greene and Johnson2014). This suggests that reduced synaptic connectivity in these regions and layers could disrupt cognitive processes in ways that contribute to the symptoms of schizophrenia. For example, deficits in memory of whether a phrase was heard or imagined, a process that involves both the medial prefrontal cortex and superior frontal cortex, could contribute to the experience of hallucinations (Brandt et al., Reference Brandt, Bergström, Buda, Henson and Simons2014; Sugimori et al., Reference Sugimori, Mitchell, Raye, Greene and Johnson2014). Reductions in synaptic density and function have also been observed in the hippocampus, and may be more prevalent in CA4 and CA3 relative to CA1 (Harrison & Eastwood, Reference Harrison and Eastwood2001). Though the implications of deficits in these particular regions are not well established, hippocampal dysfunction has been associated primarily with cognitive deficits in schizophrenia, such as verbal memory (Saykin et al., Reference Saykin, Shtasel, Gur, Kester, Mozley, Stafiniak and Gur1994). In sum, reduced synaptic density and functioning in the prefrontal cortex may contribute to reductions in gray matter and cognitive deficits observed in schizophrenia.
Deficits in interneuron functioning
Postmortem studies of schizophrenia cases have also identified reduced numbers of parvalbumin-containing GABAergic interneurons in the gray matter of the hippocampus and across the prefrontal cortex (Bennett, Reference Bennett2011). Interneurons are activated by excitatory pyramidal neurons, and in turn inhibit the activation of pyramidal neurons. The firing of interneurons promotes coordinated firing of cortical pyramidal neurons, which is critical to complex cognitive function (Chung, Fish, & Lewis, Reference Chung, Fish and Lewis2016). Reduced interneuron density could interfere with this coordination, interfering with cognitive processes based in the prefrontal cortex and hippocampus (described above). The contribution of disrupted pyramidal neuron communication to the development of schizophrenia is also supported by evidence implicating NMDA receptor dysfunction (Coyle, Tsai, & Goff, Reference Coyle, Tsai and Goff2003). NMDA receptors are glutamatergic postsynaptic membrane receptors that, when activated, promote firing of the postsynaptic neuron and help strengthen the synapse (Homayoun & Moghaddam, Reference Homayoun and Moghaddam2007). NMDA receptors are present on many neuron types, and their function is particularly important for interneurons. If postsynaptic NMDA receptors on interneurons fail to be activated, the interneuron is less likely to successfully fire on its target pyramidal neurons. This results in poor regulation of pyramidal neuron activity (e.g., excitation/inhibition imbalance), which undermines cognitive functioning (Homayoun & Moghaddam, Reference Homayoun and Moghaddam2007). The influence of NMDA receptor function on symptoms of schizophrenia is also supported by ketamine studies, which have been shown to induce psychotic symptoms by blocking NMDA receptors in the prefrontal cortex (Krystal et al., Reference Krystal, Karper, Seibyl, Freeman, Delaney, Bremner and Charney1994). Thus, reduced interneuron density and function may also contribute to the symptoms of schizophrenia.
White matter disruption
Disruption to white matter may also influence gray matter volume and contribute to the emergence of these symptoms. The speed and efficiency of communication between neurons is improved with myelination, the wrapping of fatty myelin-containing cells around axons of neurons (Davis et al., Reference Davis, Stewart, Friedman and Buchsbaum2003). Decreased white matter volume in the prefrontal cortex, particularly in the orbitofrontal region, has been associated with increased negative symptoms in individuals with schizophrenia (Sanfilipo et al., Reference Sanfilipo, Lafargue, Rusinek, Arena, Loneragan, Lautin and Wolkin2000). In addition, reduced integrity has been observed in white matter tracts throughout the brain (Lim et al., Reference Lim, Hedehus, Moseley, de Crespigny, Sullivan and Pfefferbaum1999). Deficits in certain tracts have been linked with cognitive deficits in patients, such as poorer attention and working memory associated with alterations in the cingulum bundle (Kubicki et al., Reference Kubicki, Westin, Nestor, Wible, Maier, Kikinis and Shenton2003). Alterations in myelination have been observed in clinically high-risk individuals and first episode patients as well (Witthaus et al., Reference Witthaus, Brüne, Kaufmann, Bohner, Özgürdal, Gudlowski and Juckel2008), though whether these changes are associated with onset of the disorder remains unclear (Cannon et al., Reference Cannon, Chung, He, Sun, Jacobson, van Erp and Heinssen2015).
Connection to dopamine regulation
Deficits in synaptic density, interneuron function, and myelination converge on disruption of communication between cortical and hippocampal pyramidal neurons. Cumulatively, this dysfunction may lead to downstream changes in other areas of the brain that also contribute to the symptoms of schizophrenia. For instance, increased firing of dopaminergic neurons in the striatum has been induced by disruption of pyramidal cell firing in the prefrontal cortex and hippocampus in animal models (Kim et al., Reference Kim, Rossi, Aryal, Racz, Kim, Uezu and Soderling2015; Lodge & Grace, Reference Lodge and Grace2007). Excess dopamine in the striatum is a hallmark finding in schizophrenia, and the efficacy of antipsychotic medication is attributed to countering this neurotransmitter excess (Creese, Burt, & Snyder, Reference Creese, Burt and Snyder1976). Dopamine in the striatum has been implicated in reward and salience-related signaling (Schultz, Reference Schultz1998). Excess dopamine has been associated with salience being attributed to unimportant stimuli in individuals with schizophrenia, and may contribute to the emergence of delusions (Roiser, Howes, Chaddock, Joyce, & McGuire, Reference Roiser, Howes, Chaddock, Joyce and McGuire2013). The role of the prefrontal cortex in regulating subcortical dopamine signaling further supports the theory that that disruption of coordinated communication between prefrontal cortical pyramidal neurons may play an early role in the etiology of schizophrenia.
Onset of schizophrenia
A prominent neurobiological theory of schizophrenia suggests that positive symptoms emerge when a key threshold of prefrontal and temporal cortical synaptic density is crossed during synaptic pruning in adolescence (Cannon, Reference Cannon2015; Feinberg, Reference Feinberg1982; Weinberger, Reference Weinberger1987). This theory integrates the neurobiological findings described above and may help explain the proximal onset of positive symptoms of schizophrenia in adolescence. However, the question remains as to whether psychosis results from a rapid disintegration of synaptic functioning, interneuron coordination, and/or myelination, or if deficits in these systems are present early in development and compound over time. Clarifying the timing of the emergence of these deficits can further clarify etiological pathways to psychosis and potential time points of interaction with exposure to factors such as inflammation to further increase risk for the development of the disorder.
Developmental emergence of neurobiological deficits associated with schizophrenia
Though the onset of schizophrenia typically takes place in adolescence, evidence of less severe deficits in these systems may be apparent at much earlier time points in development. Some neural abnormalities such as increased ventricular volume and poorer global network efficiency may be noted at birth among individuals at high genetic risk (Gilmore et al., Reference Gilmore, Kang, Evans, Wolfe, Smith, Lieberman and Gerig2010; Shi et al., Reference Shi, Yap, Gao, Lin, Gilmore and Shen2012). Some of those who ultimately develop schizophrenia perform worse on verbal memory, cognitive set-shifting, perceptual-motor, and timed working memory tasks in early childhood (Bearden et al., Reference Bearden, Rosso, Sanchez, Hadley, Nuechterlein and Cannon2003; Brown et al., Reference Brown, Vinogradov, Kremen, Poole, Bao, Kern and McKeague2011; Ellman, Yolken, Buka, Torrey, & Cannon, Reference Ellman, Yolken, Buka, Torrey and Cannon2009). These findings suggest that at least some individuals may exhibit deficits in these cellular mechanisms as early as prenatal development. These deficits may compound as neurodevelopment continues across childhood and adolescence, leading to increasingly severe deficits that ultimately contribute to onset of the disorder. In other cases, individuals seem to exhibit more typical cognitive development in childhood and early adolescence, then exhibit a rapid decrease in functioning in later adolescence that culminates in a psychotic episode (Cannon et al., Reference Cannon, Theo, Erp, Bearden, Loewy, Thompson and Tsuang2003; Jacobsen & Rapoport, Reference Jacobsen and Rapoport1998). It is possible that individuals who follow this symptom trajectory have subtle and/or quiescent deficits in these cellular processes that are “unmasked” by adolescent neurodevelopmental processes and/or environmental exposures that lead to a rapid decrease in neural functioning. Thus, genetic and environmental influences that are implicated in risk for schizophrenia may affect the development of these cellular mechanisms as early as the prenatal period. Certain influences may affect the development of these cellular mechanisms at distinct time points, which may result in different effects that ultimately lead to the same disorder. Understanding the influence of specific risk factors for schizophrenia, such as prenatal inflammation, on these cellular mechanisms may provide further insight into developmental pathways to schizophrenia.
Influence of inflammation on the development of implicated cellular mechanisms
Deficits in cognitive and motor development may be more apparent in individuals with schizophrenia who were exposed to prenatal inflammatory challenges compared to cases who did not experience prenatal insults (Brown et al., Reference Brown, Cohen, Harkavy-Friedman, Babulas, Malaspina, Gorman and Susser2001, Reference Brown, Vinogradov, Kremen, Poole, Deicken, Penner and Schaefer2009; Ellman et al., Reference Ellman, Yolken, Buka, Torrey and Cannon2009). This pattern suggests that inflammation may synergize with genetic risk to perturb the development of cellular mechanisms that are implicated in the emergence of schizophrenia. Rodent models of maternal immune activation have identified many changes in neural structure and behavior that parallel those observed in schizophrenia, including anatomical changes such as reductions in synaptic density between layer III pyramidal neurons in the dorsolateral prefrontal cortex (Weir et al., Reference Weir, Forghany, Smith, Patterson, McAllister, Schumann and Bauman2015), reduced gray matter volume across the cortex (Short et al., Reference Short, Lubach, Karasin, Olsen, Styner, Knickmeyer and Coe2010), and reduced myelin (Rodts-Palenik et al., Reference Rodts-Palenik, Wyatt-Ashmead, Pang, Thigpen, Cai, Rhodes and Bennett2004), as well as cognitive changes such as deficits in social interactions (Shi, Fatemi, Sidwell, & Patterson, Reference Shi, Fatemi, Sidwell and Patterson2003) and impairment in associative learning and memory tasks (Golan, Lev, Hallak, Sorokin, & Huleihel, Reference Golan, Lev, Hallak, Sorokin and Huleihel2005). In adolescents at clinical high risk who ultimately converted to psychosis, higher rates of thinning in the superior and medial frontal cortex and medial orbitofrontal cortex were correlated with both the severity of positive symptoms and levels of inflammatory markers in peripheral blood samples (Cannon et al., Reference Cannon, Chung, He, Sun, Jacobson, van Erp and Heinssen2015). Understanding the influence of inflammation on the development of these cellular mechanisms that provide the foundation for subsequent cognition and behavior could lead to more insight into etiological pathways to schizophrenia and ultimately provide novel opportunities for intervention and prevention.
To understand the influence of prenatal inflammation on these cellular mechanisms, it is important to first establish typical neurodevelopmental patterns. Next, we will review neurobiological development from the prenatal period through childhood, adolescence, and early adulthood, and consider how neural changes may relate to cognitive development across this period.
Overview of Neurobiological Development
Prenatal and neonatal development
Pyramidal neurons, interneurons, and synapses
See Figure 1 for a visual timeline of neurobiological development. The development of the CNS starts with the formation of the neural tube around the third gestational week, which splits into a series of vesicles that ultimately form the forebrain, midbrain, and hindbrain (Tau & Peterson, Reference Tau and Peterson2010). The forebrain vesicle further splits into the telencephalon (cerebral cortex, basal ganglia, and hippocampus) and diencephalon (including the thalamus and hypothalamus; Catts et al., Reference Catts, Fung, Long, Joshi, Vercammen, Allen and Shannon Weickert2013; Tau & Peterson, Reference Tau and Peterson2010). Proliferation of neural progenitor cells that will later mature into neurons begins in the dorsal and ventral regions of the telencephalon in weeks 5–6 and continues through middle gestation (~23 weeks). Cortical pyramidal neurons are generated in the dorsal (lateral ventrical) region, and GABAergic interneurons originate in the ventral (early basal ganglia/subventricular) region (Catts et al., Reference Catts, Fung, Long, Joshi, Vercammen, Allen and Shannon Weickert2013; Tau & Peterson, Reference Tau and Peterson2010). Neuronal migration peaks between the start of the second trimester (~13 weeks) and about 20 weeks, following a subcortical to cortical pattern (Catts et al., Reference Catts, Fung, Long, Joshi, Vercammen, Allen and Shannon Weickert2013; Tau & Peterson, Reference Tau and Peterson2010). Neurons originating in the dorsal ventricular zone are typically guided by radial glia to their designated location through the developing cortex, whereas interneurons migrate in parallel to the developing cortex before reaching their destination (Tau & Peterson, Reference Tau and Peterson2010). Neuronal migration to the cortex gives rise to the preplate, a transient structure that forms the first identifiable cortical layer (Tau & Peterson, Reference Tau and Peterson2010). By weeks 7–8, the formation of the cortical plate splits the preplate into the subplate and the marginal zone. The subplate grows rapidly in middle gestation (weeks 18–22) and serves as a placeholder for early connections between cortical and subcortical neurons. By the beginning of the third trimester (~26 weeks), the subplate dissolves and the marginal zone (cortical layer I) and cortical plate (cortical layers II–VI) form the basis of the mature organizational structure of the cortex. Neuron proliferation and migration end at about 30 weeks with the completion of association cortices and commissural regions (de Graaf-Peters & Hadders-Algra, Reference de Graaf-Peters and Hadders-Algra2006; Tau & Peterson, Reference Tau and Peterson2010), though accumulating evidence suggests that interneuron proliferation and migration from the ventral subventricular zone may continue at low levels into adulthood (Catts et al., Reference Catts, Fung, Long, Joshi, Vercammen, Allen and Shannon Weickert2013).
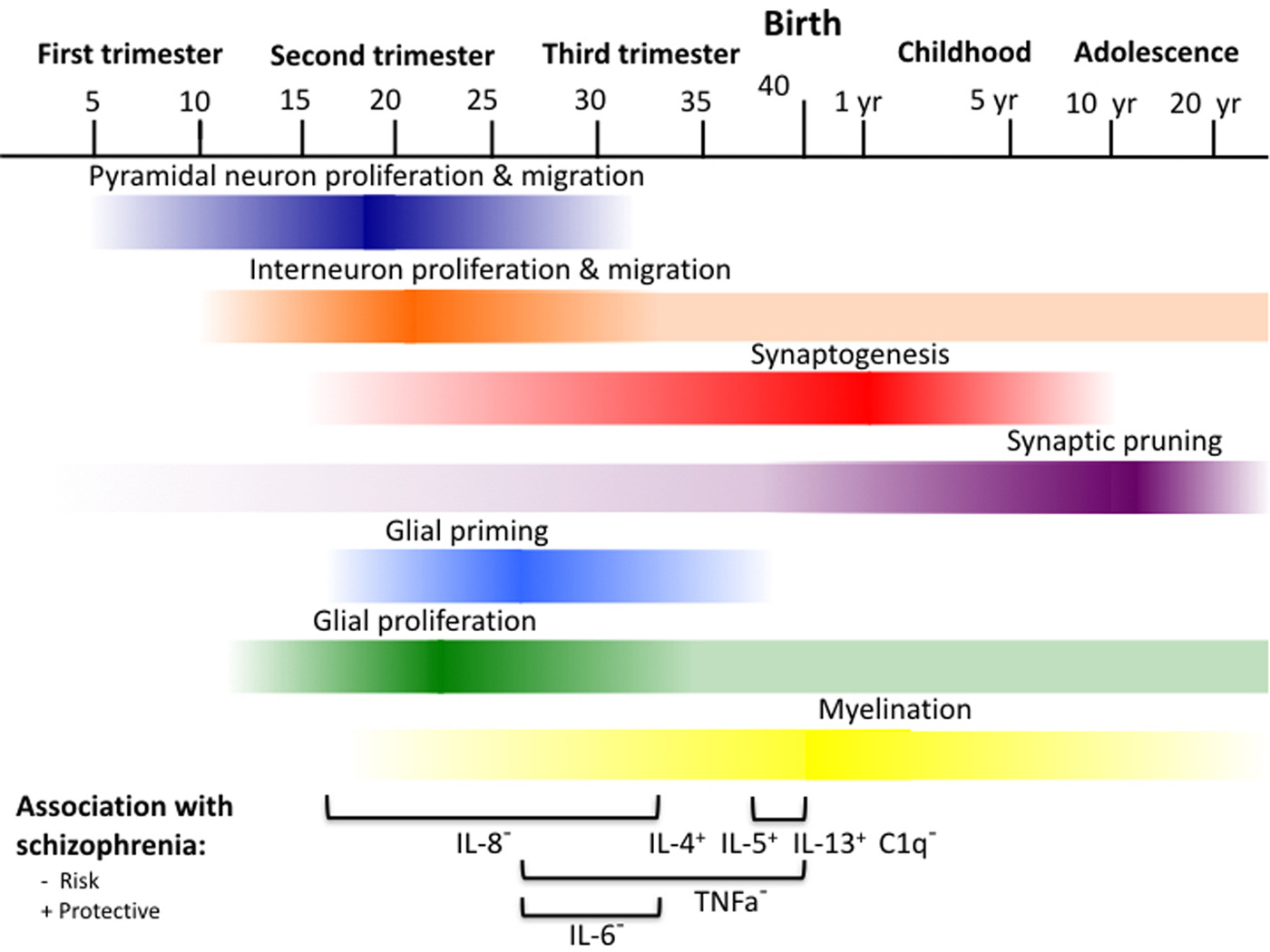
Figure 1. (Color online) Timeline of neurodevelopmental processes and specific prenatal cytokine and complement elevations associated with schizophrenia. IL-8, 13–32 weeks; IL–6, 26–32 weeks; TNFa, 26–42 weeks; IL-4, IL5, IL-13, C1q, 37–42 weeks.
Maturation of neurons, including proper alignment of cells and neuronal communication via axon and dendritic outgrowth and the formation of synaptic connections, begins around 16 to 24 weeks as migration is completed (Catts et al., Reference Catts, Fung, Long, Joshi, Vercammen, Allen and Shannon Weickert2013; Tau & Peterson, Reference Tau and Peterson2010). Around this time, cells in regions such as the thalamus, brainstem, and nucleus accumbens send preliminary axonal projections to form synapses with cortical placeholder neurons in the subplate (Tau & Peterson, Reference Tau and Peterson2010). These connections are refined and transferred to enduring neurons in the cortical plate around 24–28 weeks gestation as the subplate dissolves, paving the way for more sophisticated cortical development (Tau & Peterson, Reference Tau and Peterson2010). Neural cells begin to organize into mini columns, and cortical layers composed of different cell types and connections with other regions start to form. Evidence of cortical layering is first present in primary sensory and motor regions starting around week 25 (Tau & Peterson, Reference Tau and Peterson2010). In gestational weeks 24–34, cortical neurons first form synaptic connections nonspecifically with other local cells in the marginal zone. Ultimately, these synaptic connections become cortical layer I, the most superficial cortical layer (Tau & Peterson, Reference Tau and Peterson2010). Cortical pyramidal neurons then begin to send axonal projections to ipsilateral and contralateral cortical regions (layer III) and integrate projections from other regions such as the thalamus and brainstem (layer IV; Tau & Peterson, Reference Tau and Peterson2010). By the late third trimester, pyramidal neurons begin to preferentially form local synaptic connections with other pyramidal neurons in different layers of the same column of cells (layers V and IV) and with other pyramidal neurons in the same layer (layer II; Tau & Peterson, Reference Tau and Peterson2010). By about week 32, the foundation of cortical layers is present throughout the cortex, including integration of projections of all major neurotransmitter systems and a variety of neuronal (i.e., pyramidal and interneuron) and glial cell types (Tau & Peterson, Reference Tau and Peterson2010).
Once the foundation of cortical layering is established, rates of synaptic formation increase rapidly. Rates of growth in synaptic density peak at a rate of almost 40,000 new synapses formed every second from the third trimester through the first neonatal year (de Graaf-Peters & Hadders-Algra, Reference de Graaf-Peters and Hadders-Algra2006; Tau & Peterson, Reference Tau and Peterson2010). In the third trimester and first few neonatal months, synaptic density grows at a particularly rapid rate in primary sensory and motor regions (Huttenlocher & Dabholkar, Reference Huttenlocher and Dabholkar1997), and connections formed are primarily between local neurons (e.g., cortical layers I and II; Tau & Peterson, Reference Tau and Peterson2010). Refinement of synaptic connections via pruning of competing synapses co-occurs at a low rate with synaptic formation in early development, but synaptic outgrowth dominates at this stage of development (de Graaf-Peters & Hadders-Algra, Reference de Graaf-Peters and Hadders-Algra2006).
Glia
Two subtypes of glial cells follow similar patterns of development as neurons. Like neurons, both astroctyes and oligodendrocytes are derived from neural progenitor cells and are generated in that order once neurogenesis for neurons destined for a particular neural region has been completed (Eroglu & Barres, Reference Eroglu and Barres2015). Proliferation of astrocytes and oligodendrocytes is at its greatest rate from about 20 to 40 weeks gestation, but begins as early as 10 weeks and continues after birth for oligodendrocytes (de Graaf-Peters & Hadders-Algra, Reference de Graaf-Peters and Hadders-Algra2006). Astrocytes tend to localize to synapses and play key roles in supporting synaptic formation and plasticity (Eroglu & Barres, Reference Eroglu and Barres2015). The timing of gliogenesis coincides with synaptogenesis, likely due to the important role of astrocytes in this process (Semple, Blomgren, Gimlin, Ferriero, & Noble-Haeusslein, Reference Semple, Blomgren, Gimlin, Ferriero and Noble-Haeusslein2013). Oligodendrocytes form myelin in their mature state and wrap around axons, increasing the speed of signal transmission within neurons (Eroglu & Barres, Reference Eroglu and Barres2015). Myelination follows similar patterns as synaptic growth, beginning in the precursor to the basal ganglia around 14 weeks, in the subcortical striatum around 28 weeks, and in the cortex around 35 weeks (de Graaf-Peters & Hadders-Algra, Reference de Graaf-Peters and Hadders-Algra2006). Rates of myelination peak in the third trimester and first 3 months of life (Catts et al., Reference Catts, Fung, Long, Joshi, Vercammen, Allen and Shannon Weickert2013). For instance, whole-brain volume that contains mature white matter increases from 1% to 5% between gestational weeks 36 and 40, and continues after birth (Catts et al., Reference Catts, Fung, Long, Joshi, Vercammen, Allen and Shannon Weickert2013).
One class of glia, microglia, does not originate from neural progenitor cells but instead from immune progenitors outside of the brain (Deverman & Patterson, Reference Deverman and Patterson2009). Immature microglia are thought to migrate into the CNS very early in neurodevelopment, possibly in response to markers of programmed cell death that begin starting at 7 weeks gestation and continue through the first trimester (de Graaf-Peters & Hadders-Algra, Reference de Graaf-Peters and Hadders-Algra2006). Microglia seem to be in an activated state during the late second and early third trimester through the early postnatal period, and play a critical role in promoting survival of neurons as well as shaping synaptic strength and density (Bilbo & Schwarz, Reference Bilbo and Schwarz2009). Microglia are thought to mature into their quiescent adult form within the early neonatal period and only take an active form in response to later immune challenges (Bilbo & Schwarz, Reference Bilbo and Schwarz2009).
Neonatal neural structure
By the end of prenatal development, much of the cellular and structural framework of the brain has been established. This progress can also be observed with noninvasive structural and functional scans of the developing human brain. For instance, fetal magnetic resonance imaging scans have shown that gyri and sulci appear in the thickening developing cortex as synaptogenesis accelerates during the third trimester, and total brain volume increases nearly threefold during this period (Tau & Peterson, Reference Tau and Peterson2010). Functionally, it seems that basic visual, sensorimotor, and auditory processing circuits are functional at term birth (39–44 weeks; Fransson, Ulrika, Blennow, & Lagercrantz, Reference Fransson, Ulrika, Blennow and Lagercrantz2011), and possibly a preliminary default mode network connecting prefrontal and parietal regions as well (Tau & Peterson, Reference Tau and Peterson2010). This provides the foundation for the emergence of basic sensorimotor and visual functioning at the time of birth (Tau & Peterson, Reference Tau and Peterson2010). Continued growth and refinement of the neural framework during the postnatal period gives rise to increasingly complex cognition and behavior.
Childhood and adolescent neural and cognitive development
The first few postnatal years are characterized by rapid increases in the size of the brain. At 2–4 weeks after birth, the brain is 36% of adult size. By the end of the first year, the brain is 70% of adult size, and is about 80% of the adult size by age 2 (Knickmeyer et al., Reference Knickmeyer, Gouttard, Kang, Evans, Wilber, Smith and Gilmore2008). This increase in volume is greatest in the cerebellum, followed by subcortical areas and the cerebral cortex (Knickmeyer et al., Reference Knickmeyer, Gouttard, Kang, Evans, Wilber, Smith and Gilmore2008), and is thought to be driven by the expansion of synaptic connections in gray matter, glia, and myelination (Tau & Peterson, Reference Tau and Peterson2010). In the cortex, growth may be particularly driven by dendritic outgrowth and synaptic formation of both pyramidal neurons and interneurons (Mrzljak, Uylings, van Eden, & Judas, Reference Mrzljak, Uylings, van Eden and Judas1990). Synaptic density peaks at different times in regions across the brain in back to forward and subcortical to cortical patterns, starting with primary sensory regions (~3 months) and followed by association regions and the prefrontal cortex (~15 months; Huttenlocher & Dabholkar, Reference Huttenlocher and Dabholkar1997). Synaptic connections between local neurons (e.g., cortical layers I and II) form earlier than connections between more distant neurons (e.g., cortical layers III, IV, V, and VI) in the first 2 to 3 years of life (Tau & Peterson, Reference Tau and Peterson2010). Myelination also occurs at rapid rates within the first few years of life, beginning with proximal connections and projections in sensory and motor regions and expanding to distal connections and association fibers and regions (Lenroot & Giedd, Reference Lenroot and Giedd2006). By three years of age, myelination of the neocortex reaches 95% of adult levels (Kang et al., Reference Kang, Kawasawa, Cheng, Zhu, Xu, Li and Sestan2011). Markers of interneuron subtypes, including parvalbumin and cholecystokinin, increase across the first 5 years of life, suggesting that interneuron maturation takes place during this period as well (Catts et al., Reference Catts, Fung, Long, Joshi, Vercammen, Allen and Shannon Weickert2013).
Though the first few years of neurodevelopment are characterized by rapid growth, refinement of systems that become functional early in life also begins to take place. In the first 2 years of life, sensory and motor regions and circuits complete the majority of their growth and undergo substantial refinement (Levitt, Reference Levitt2003; Tau & Peterson, Reference Tau and Peterson2010). This growth and refinement is paralleled by improvements in infant sensory and motor functioning and ability. For example, at the time of birth, infants are capable of basic attentional processes such as tracking slowly moving stimuli and fixating on objects of interest (Johnson, Reference Johnson1990). These processes exhibit improvement across the first few months of life, such as increased sensitivity to the nasal visual field, anticipatory tracking of moving objects, and longer looking times to objects that violate implicit predictions about movement. These changes parallel maturation of regions including the primary visual cortex, superior colliculus, and basal ganglia (Johnson, Reference Johnson1990). These basic visual attention processes and the implicated regions of the brain complete maturity within the first year of life (Johnson, Reference Johnson1990), with improvements in resting functional connectivity of visual regions improving through the second postnatal year (Lin et al., Reference Lin, Zhu, Gao, Chen, Toh, Styner and Gilmore2008). The emergence of these visual abilities suggests that preliminary circuitry supporting spatial “where” orientation (parietal–thalamic circuit), timing “when” orientation (such as cerebellar–cortical circuitry), inhibitory “what” orientation (frontostriatal), and reinforcement learning (frontolimbic) may be present early in life (Nigg & Casey, Reference Nigg and Casey2005). Coordination of these processes is thought to give rise to the capacity to move toward a goal by planning one's behavior, often referred to as higher level cognition or executive function (Shallice, Reference Shallice1982). Though initial evidence of these systems underlying cognition emerges in the first year of life, the ability to perform complex tasks such as focusing on a relevant object in the presence of competing objects does not emerge until middle childhood (Herschkowitz, Reference Herschkowitz2000; Nigg & Casey, Reference Nigg and Casey2005). This suggests that further maturation of these circuits and underlying regions that communicate with the visual system is needed to support more advanced cognitive development.
Neural growth and refinement continues during early and middle childhood. Broadly, robust synaptic formation continues at a slower rate until approximately 10 years of age (Catts et al., Reference Catts, Fung, Long, Joshi, Vercammen, Allen and Shannon Weickert2013). Outgrowth of projections destined for distant neurons (e.g., cortical layers III, IV, V, and VI) increase (Tau & Peterson, Reference Tau and Peterson2010), as do myelination and interneuron proliferation and migration (Catts et al., Reference Catts, Fung, Long, Joshi, Vercammen, Allen and Shannon Weickert2013). Association and distal connections are increasingly myelinated, giving rise to increased connectivity between the same regions across hemispheres followed by long-range connections between regions in the same hemisphere (Lenroot & Giedd, Reference Lenroot and Giedd2006; Tau & Peterson, Reference Tau and Peterson2010). These processes contribute to gray matter volumes peaking in subcortical to cortical and posterior to anterior patterns, including the caudate around 7–10 years and regions of the prefrontal, parietal, and temporal lobes closest to primary motor and sensory regions around 8–12 years (Gogtay et al., Reference Gogtay, Giedd, Lusk, Hayashi, Greenstein, Vaituzis and Thompson2004; Lenroot & Giedd, Reference Lenroot and Giedd2006). Reductions in volume may reflect synaptic refinement (Glantz & Lewis, Reference Glantz and Lewis2000) and/or increasing myelination (Catts et al., Reference Catts, Fung, Long, Joshi, Vercammen, Allen and Shannon Weickert2013) within these regions. Refinement of these regions supports the maturation of abilities such as language and attention and improvements in inhibitory frontostriatal “what,” frontocerebellar “when,” and frontoparietal working memory circuitry (Nigg & Casey, Reference Nigg and Casey2005). For example, cerebellar maturation gives rise to improved fine motor control and visuomotor coordination across childhood, and improved coordination of cerebellar and dorsal prefrontal activity supports learning of novel skills and improved error monitoring (Diamond, Reference Diamond2000). Children activate frontoparietal circuitry during simple working memory tasks, suggesting that this system is functional in middle childhood. However, when working memory load is increased with the introduction of distractors or requiring mental manipulation of information, children tend to additionally recruit regions such as the caudate and insula rather than increasing frontoparietal circuitry activity like adults (Tau & Peterson, Reference Tau and Peterson2010). This suggests that cognition in childhood may be particularly sensitive to interference, which may reflect the lack of maturity of cortical association areas that integrate information from a variety of regions (Casey, Tottenham, Liston, & Durston, Reference Casey, Tottenham, Liston and Durston2005; Tau & Peterson, Reference Tau and Peterson2010). To achieve adult levels of cognitive functioning, continued maturation of association regions and improved efficiency of neural circuitry is needed.
In late childhood, adolescence, and early adulthood, higher order association areas such as the dorsolateral prefrontal cortex and lateral temporal cortex exhibit reductions in volume (Gogtay et al., Reference Gogtay, Giedd, Lusk, Hayashi, Greenstein, Vaituzis and Thompson2004). These association areas integrate information from regions and circuits such as sensorimotor, language, attention, and reward functioning. Maturation of association regions is thought to contribute to the development of more advanced cognitive functions such as cognitive control, the ability to pursue and monitor desired behaviors while inhibiting competing behaviors (Tau & Peterson, Reference Tau and Peterson2010). This period of development is also characterized by pruning of connections between neurons that are spatially close but functionally distinct, as well as strengthening of long-range connectivity between regions in a given circuit. This refinement of neural connectivity, in combination with increasing myelination of association tracks, results in improved efficiency and recruitment of neural circuits that supports cognition (Tau & Peterson, Reference Tau and Peterson2010). An example of this is changes in go/no-go performance across childhood and adolescence, which relies on inhibitory frontostriatal circuits that include late-maturing regions such as the dorsolateral prefrontal cortex (Nigg & Casey, Reference Nigg and Casey2005). Children tend to recruit a variety of regions outside of the frontostriatal circuit to complete the task, suggesting that this circuit and underlying regions are not yet efficient enough to support inhibition on their own. However, adolescents strongly recruit a smaller number of cortical regions and perform better on the task (Durston et al., Reference Durston, Davidson, Tottenham, Galvan, Spicer, Fossella and Casey2006). Improved performance on this task is observed from mid childhood to early adulthood, and is associated with increasing levels of frontostriatal myelin (Liston et al., Reference Liston, Watts, Tottenham, Davidson, Niogi, Ulug and Casey2006).
Broadly, adolescent improvements in neural efficiency parallel improvements in cognitive control, including shifting between sets of information, working memory capacity, and inhibition of competing responses (Huizinga, Dolan, & van der Molen, Reference Huizinga, Dolan and van der Molen2006). These advances are reflected in increasing capacity to switch strategies rather than persevere when problem solving, solve more complex problems by planning ahead and using more efficient strategies, and respond quickly and accurately (Huizinga et al., Reference Huizinga, Dolan and van der Molen2006; Levin et al., Reference Levin, Culhane, Hartmann, Evankovich, Mattson, Harward and Fletcher1991). However, it is important to note that neural and cognitive development from childhood to adolescence and into adulthood is not always linear. For instance, adolescents are more impulsive in response to appetitive cues compared to children and adults. This is suggestive of increased reward sensitivity during this period of development, which may relate to the late maturation of the dorsolateral prefrontal cortex. This pattern may explain higher rates of risk taking during adolescence compared to childhood and adulthood (Somerville, Hare, & Casey, Reference Somerville, Hare and Casey2011). Continued refinement and myelination of association areas and circuitry across adolescence into early adulthood is needed to reach full maturation of the brain and cognition.
Summary
Prenatal development sets the framework for neural structure and function. In general, neurodevelopment follows a subcortical to cortical and sensorimotor to association region pattern. The first half of pregnancy is characterized by neuronal and glial proliferation and migration. Synaptic formation, particularly between local neurons, and myelination contributes to rapid growth toward the middle and end of pregnancy.
Substantial portions of neurodevelopment are completed by birth, and are well established by the second or third year of life. Maturation of regions and circuits via synaptic refinement and myelination begins in sensorimotor regions in the neonatal period and continues throughout childhood and adolescence into early adulthood. This is reflected in protracted development (e.g., decreases in gray matter and increase in white matter) in the frontal, parietal, and temporal association cortices and associated neural circuits and parallels the emergence and refinement of complex cognitive processes, such as attention orienting, timing, working memory, and inhibitory control.
Alterations to prenatal neural development have the potential to affect neural and cognitive development across the life span. As prenatal exposure to inflammation has been implicated in risk for schizophrenia, and inflammatory proteins have been increasingly implicated in neurodevelopment (Ratnayake et al., Reference Ratnayake, Quinn, Walker and Dickinson2013), it is possible that prenatal inflammation affects neurodevelopment in ways that increase the risk of later developing schizophrenia in vulnerable individuals. Next, we will review two microglial-secreted immune signaling pathways, cytokines and complement, that may mediate the observed associations between maternal immune activation and risk for schizophrenia. We identify ways in which these immune proteins may be involved in typical prenatal neurodevelopment, and discuss specific markers that have been implicated in risk for schizophrenia. We consider ways that alterations in these proteins at different time points during prenatal development, in combination with genetic vulnerabilities, could potentially influence development to increase risk for schizophrenia.
Role of Immune Proteins in Neurodevelopment and Schizophrenia Risk
Cytokines
One immune pathway implicated in risk for schizophrenia is cytokines. Cytokines are signaling proteins produced by both immune and other cell types that play a key regulatory role in the initiation and maintenance of the immune response (Deverman & Patterson, Reference Deverman and Patterson2009). Cytokine levels are elevated in response to markers of infection and contribute to the recruitment and activation of immune cells (Deverman & Patterson, Reference Deverman and Patterson2009; Meyer et al., Reference Meyer, Feldon and Yee2009). Cytokines are often broadly categorized as pro-inflammatory (e.g., IL-1β, IL-2, IL-6, IL-8, TNFα, and IFNγ) or anti-inflammatory (e.g., IL-4, IL-5, IL-10, and TGFβ; Meyer, Feldon, & Yee, Reference Meyer, Feldon and Yee2009); however, some cytokines can function differently depending on the context (e.g., IL-10; Meyer et al., Reference Meyer, Murray, Urwyler, Yee, Schedlowski and Feldon2008). In both the developing and adult CNS, cytokines are primarily produced by microglia and astrocytes (Deverman & Patterson, Reference Deverman and Patterson2009). Most cytokines are not thought to cross the intact blood–brain barrier. However, peripheral inflammation can be transduced to the CNS via a secondary immune response at the blood–brain barrier that triggers a neuroinflammatory response from glial cells (Bergink et al., Reference Bergink, Gibney and Drexhage2014). A similar process is thought to occur at the placenta, and elevated maternal cytokine levels have been shown to broadly alter cytokine levels in the placenta, amniotic fluid, and fetal brain (Urakubo, Jarskog, Lieberman, & Gilmore, Reference Urakubo, Jarskog, Lieberman and Gilmore2001).
Prenatal cytokine exposure and risk for schizophrenia
A variety of prenatal risk factors for schizophrenia have been linked with increases in maternal and fetal cytokine levels, such as viral infections with IL-6 (Cunningham, Campion, Teeling, Felton, & Perry, Reference Cunningham, Campion, Teeling, Felton and Perry2007), bacterial infections with IL-1β and TNFα (Cai et al., Reference Cai, Pan, Pang, Evans and Rhodes2000), and hypoxic/ischemic birth insults with IL-1β and IL-2 (Girard, Kadhim, et al., Reference Girard, Kadhim, Larouche, Roy, Gobeil and Sébire2008; Girard, Larouche, et al., Reference Girard, Larouche, Kadhim, Rola-Pleszczynski, Gobeil and Sébire2008). Only a handful of studies have examined whether elevations in individual cytokines during pregnancy are associated with risk for schizophrenia in human samples. Epidemiologic studies examining alterations in maternal cytokine levels during pregnancy in association with risk for schizophrenia have identified second/third trimester elevations in maternal IL-8 (and not IL-1β, IL-6, or TNFα; Brown, Hooton, et al., Reference Brown, Hooton, Schaefer, Zhang, Petkova, Babulas and Susser2004; Ellman et al., Reference Ellman, Deicken, Vinogradov, Kremen, Poole, Kern and Brown2010), third trimester elevations in IL-6 for males and low levels of TNFα for females (and not IL-1β, IL-8, or IL-10; Goldstein et al., Reference Goldstein, Cherkerzian, Seidman, Donatelli, Remington, Tsuang and Buka2014), and elevations in TNFα (and not IL-1β, IL-2, IL-6, or IL-8) at the time of delivery (Buka, Tsuang, Torrey, Klebanoff, Wagner, et al., Reference Buka, Tsuang, Torrey, Klebanoff, Wagner and Yolken2001; Figure 1). In addition, elevations in a combined measure of maternal IL-4, IL-5, and IL-13 at birth have been identified as a protective factor (Allswede, Buka, Yolken, Torrey, & Cannon, Reference Allswede, Buka, Yolken, Torrey and Cannon2016). Levels of cytokines from neonatal blood samples have not consistently been associated with later risk for schizophrenia. This suggests that earlier prenatal development may be a particularly vulnerable time frame for cytokine-mediated influences on neurodevelopment that increase risk for schizophrenia (Nielsen et al., Reference Nielsen, Agerbo, Skogstrand, Hougaard, Meyer and Mortensen2014).
Role of cytokines in typical neurodevelopment
Rodent studies suggest a role for cytokines in most aspects of prenatal neurodevelopment throughout the brain, including neuronal and glial migration and differentiation, synaptic maturation, and myelination (Depino, Reference Depino2013; Ratnayake et al., Reference Ratnayake, Quinn, Walker and Dickinson2013). Both pro- and anti-inflammatory cytokines have been observed in the brain as early as 5 weeks gestation and exhibit different patterns of elevation across early gestation (Dziegielewska et al., Reference Dziegielewska, Moller, Potter, Ek, Lane and Saunders2000; Mousa, Seiger, Kjaeldgaard, & Bakhiet, Reference Mousa, Seiger, Kjaeldgaard and Bakhiet1999). Certain cytokines appear to play specific roles in distinct neurodevelopmental processes. For example, IL-6, but not IFNγ, has been shown to influence the generation of neural progenitor cells in the adult subventricular zone, and may play a similar role in early stages of neurogenesis (Gallagher et al., Reference Gallagher, Norman, Woodard, Yang, Gauthier-Fisher, Fujitani and Miller2013). IL-6 has also been implicated in mediating the transition from neurogenesis to astrogenesis (Semple et al., Reference Semple, Blomgren, Gimlin, Ferriero and Noble-Haeusslein2013). IL-1β has been shown to guide the migration of cortical neurons (Ma et al., Reference Ma, Li, Zhang, Yang, Zhu, Yuan and Jiang2014), and may be particularly effective at promoting the maturation of midbrain neural progenitor cells to a dopaminergic phenotype and regulating their survival (Meyer et al., Reference Meyer, Feldon and Yee2009). IL-1β, IL-6, TNFα, and IFNγ have been implicated in regulation of cortical neuron dendrite development (Barish, Mansdorf, & Raissdana, Reference Barish, Mansdorf and Raissdana1991; Gilmore et al., Reference Gilmore, Fredrik Jarskog, Vadlamudi and Lauder2004), and TNFα has been shown to regulate expression of AMPA receptors at synapses (Eroglu & Barres, Reference Eroglu and Barres2015). IL-1β has also been implicated in regulating proliferation and maturation of oligodendrocytes and myelin levels (Cai, Lin, Pang, & Rhodes, Reference Cai, Lin, Pang and Rhodes2004; Cammer & Zhang, Reference Cammer and Zhang1999; Favrais et al., Reference Favrais, van de Looij, Fleiss, Ramanantsoa, Bonnin, Stoltenburg-Didinger and Gressens2011). Though certain cytokines, such as IL-6, appear to be implicated in many of these early neurodevelopmental processes, other cytokines are present during this period as well and have been studied less extensively. For example, similar elevation patterns in the first 10 weeks of gestation to IL-6 have been observed in IL-1β, TNFα, IFNγ, IL-4, and IL-10 (Mousa et al., Reference Mousa, Seiger, Kjaeldgaard and Bakhiet1999), suggesting that these cytokines could also potentially play a role in early neurodevelopment. These studies suggest that cytokines play specific roles in the maturing brain and that disruption of individual cytokines may have different effects on development.
Potential influence of cytokine elevations linked with schizophrenia risk on neurodevelopment
Pro-inflammatory IL-8 in mid-pregnancy
Elevations in maternal IL-8 during the second and third trimesters have been associated with risk for schizophrenia (Brown, Hooton, et al., Reference Brown, Hooton, Schaefer, Zhang, Petkova, Babulas and Susser2004). In the immune system, IL-8 is known to play a key role in attraction and activation of a subset of immune cells called neutrophils (Brown, Hooton, et al., Reference Brown, Hooton, Schaefer, Zhang, Petkova, Babulas and Susser2004). IL-8 may also play a role in placental function throughout pregnancy and the induction of labor (Brown, Hooton, et al., Reference Brown, Hooton, Schaefer, Zhang, Petkova, Babulas and Susser2004). However, specific roles for IL-8 in neurodevelopmental processes have not yet been elucidated (Brown, Hooton, et al., Reference Brown, Hooton, Schaefer, Zhang, Petkova, Babulas and Susser2004). During the late second and early third trimesters, processes such as pyramidal neuron and interneuron proliferation and migration are starting to slow down, astrocyte and oligodendrocyte formation are speeding up, and processes like synaptic formation and myelination in deeper and posterior regions of the brain are beginning (Figure 1). This suggests that elevations in maternal IL-8 may have an effect on one or more of these processes in the fetal brain. Some insight into the role of maternal IL-8 on neurodevelopment may be gained from a study examining the association between IL-8 exposure during this period of prenatal development and structural gray matter changes in adulthood among schizophrenia cases (Ellman et al., Reference Ellman, Deicken, Vinogradov, Kremen, Poole, Kern and Brown2010). Cases with higher levels of second/third trimester maternal IL-8 exhibited higher volumes of cerebrospinal fluid (CSF) in the ventricles and exhibited lower volumes in the left entorhinal cortex and right posterior cingulate cortex compared to cases with lower levels of IL-8. Decreased cortical volume suggests that maternal IL-8 levels could plausibly be associated with reduced synaptic density, neuronal migration and survival, and/or myelination in these regions. These regions are implicated in memory and communicate with one another as well as with the prefrontal cortex (Miller & Cohen, Reference Miller and Cohen2001). Memory deficits are observed along with other broader cognitive deficits in schizophrenia (Fioravanti et al., Reference Fioravanti, Carlone, Vitale, Cinti and Clare2005), and deficits in source memory may contribute to the emergence of delusions and hallucinations (Cannon, Reference Cannon2015). If cases with higher maternal IL-8 levels in pregnancy exhibit greater deficits in general memory and/or source compared to cases with lower IL-8 levels, this would suggest that high maternal IL-8 levels may be associated with these particular aspects of the disorder. Further research is needed to specify the role that maternal IL-8 may have on typical neurodevelopment in the fetus, and how alterations in maternal IL-8 levels might influence neurodevelopmental processes such as synaptic pruning or interneuron function.
Pro-inflammatory IL-6 and TNFα in late pregnancy
Elevations in maternal IL-6 and TNFα in the third trimester have been associated with risk for schizophrenia. During this period of neonatal development, processes such as synaptic formation and astrocyte and oligodendrocyte formation and maturation are starting to hit peak rates, particularly in sensorimotor regions (Figure 1). Different effects of maternal IL-6 and TNFα levels on risk for schizophrenia based on gender may reflect interactions between cytokines and sex hormones (for further discussion, see Goldstein et al., Reference Goldstein, Cherkerzian, Seidman, Donatelli, Remington, Tsuang and Buka2014). Both IL-6 and TNFα have been shown to inhibit dendrite development on cortical neurons in rats at high concentrations. TNFα has also been shown to have this effect at low concentrations (Gilmore et al., Reference Gilmore, Fredrik Jarskog, Vadlamudi and Lauder2004). It is important to note that maternal cytokine elevations typically index general inflammation in the fetal brain, but not necessarily the same cytokines (see “Correspondence Between Maternal and Fetal Immune Signaling” section below for further discussion). However, it is possible that high levels of maternal IL-6 in the third trimester, low levels of TNFα during the third trimester, and high levels of TNFα at delivery are all associated with decreased synaptic formation in the fetal brain. Depending on the timing of the insult, synaptic density of specific regions could be influenced differently. Synaptic density first peaks in visual and auditory cortices a few months after birth, and peaks in the prefrontal cortex at approximately 15 months (Huttenlocher & Dabholkar, Reference Huttenlocher and Dabholkar1997). This suggests that synapses are being formed in most regions of the brain during late gestation, with highest rates in primary sensory regions. If maternal IL-6 and TNFα lead to changes in the fetal brain that are sufficient to disrupt synaptic formation on their own, alterations in maternal IL-6 and TNFα levels at this time could plausibly affect synaptic density in all regions of the brain. If changes in the fetal brain associated with maternal IL-6 and TNFα are not sufficient to disrupt synapse formation on their own but interact with specific cell types, only certain regions may be affected. Maternal IL-6 and TNFα that induce parallel changes in the fetal brain may also exert specific influences on other neurobiological systems. For example, elevations in fetal TNFα may interfere with oligodendrocyte maturation. In cell cultures, administration of TNFα disrupts maturation of oligodendrocytes, but not the survival of precursors or immature oligodendrocytes (Cammer & Zhang, Reference Cammer and Zhang1999). These findings suggest that increases in fetal TNFα in the third trimester and at delivery may disrupt myelination, but not oligodendrocyte cell density. IL-6 has been implicated in a variety of neurodevelopmental processes, including regulation of neural progenitor cells (Gallagher et al., Reference Gallagher, Norman, Woodard, Yang, Gauthier-Fisher, Fujitani and Miller2013) and their differentiation into distinct neural and glial types (Semple et al., Reference Semple, Blomgren, Gimlin, Ferriero and Noble-Haeusslein2013). In adult neurons, elevations in IL-6 have been associated with dysfunction of GABAergic parvalbumin-containing inhibitory interneurons, which may be mediated via increases in oxidative stress (Behrens, Ali, & Dugan, Reference Behrens, Ali and Dugan2008). In sum, elevations in maternal TNFα and IL-6 in late prenatal development may interfere with many major developmental processes, including synaptic formation and glial maturation.
Anti-inflammatory IL-4, IL-5, and IL-13 at birth
There is a paucity of evidence linking anti-inflammatory cytokines to specific neurodevelopmental processes. However, there is some suggestion that these cytokines may exert influence on microglial priming. Microglia transition into an active adult form in the late second to early third trimester, which suggests that this time period may be vulnerable for glial programming and function (Bilbo & Schwarz, Reference Bilbo and Schwarz2009). Microglia can be induced into two different phenotypes: M1, which is broadly pro-inflammatory and produces primarily TNFα and IL-6, and M2, which is broadly anti-inflammatory and produces primarily IL-4, IL-10, IL-13, and TGFβ (Ponomarev, Veremeyko, & Weiner, Reference Ponomarev, Veremeyko and Weiner2013). The M1 phenotype is induced by pro-inflammatory cytokines such as IFNγ, and the M2 phenotype is induced by IL-4 and IL-13 levels. This suggests that elevated levels of IL-4, IL-5, and IL-13 during the prenatal period could protect against the development of schizophrenia by “priming” glial cells for anti-inflammatory responses (Allswede et al., Reference Allswede, Buka, Yolken, Torrey and Cannon2016). Substantial evidence suggests that exposure to pro-inflammatory cytokines during prenatal development can either sensitize or suppress microglial production of cytokines in response to immune challenges later in life (Bilbo & Schwarz, Reference Bilbo and Schwarz2009; Hodyl, Krivanek, Lawrence, Clifton, & Hodgson, Reference Hodyl, Krivanek, Lawrence, Clifton and Hodgson2007; Lasala & Zhou, Reference Lasala and Zhou2007). The association between IL-4, IL-5, and IL-13 levels and protection against development of schizophrenia was observed at the time of delivery in humans (Allswede et al., Reference Allswede, Buka, Yolken, Torrey and Cannon2016), which is later than the proposed vulnerability period for microglial priming in rodent models. However, it is possible that relatively low levels of these cytokines at delivery reflect lower levels earlier in pregnancy as well. Further research in rodent and human samples is needed to assess the potential role of anti-inflammatory signaling on microglial priming and other aspects of neurodevelopment.
Potential interactions between cytokines and genetic vulnerability to schizophrenia
Elevations in maternal cytokines have the potential to influence neurobiological systems that are implicated in schizophrenia. However, as most individuals exposed to prenatal inflammation do not develop schizophrenia, these exposures are likely not sufficient to cause schizophrenia (Mittal et al., Reference Mittal, Ellman and Cannon2008). In addition, the broad role for cytokines in neurodevelopment throughout the brain is inconsistent with patterns of specific deficits observed in the disorder, such as lower synaptic density in specifically layer III of the dorsolateral prefrontal cortex (Garey et al., Reference Garey, Ong, Patel, Kanani, Davis, Mortimer and Hirsch1998). This pattern suggests that elevations in cytokines may increase risk for schizophrenia by interacting with a variety of other vulnerability factors, such as genetic variants, that in aggregate influence certain aspects of neurodevelopment that increase risk for the development of psychosis later in life. One such interaction may be with genetic variants that have been implicated in schizophrenia, including disrupted-in-schizophrenia-1 (DISC1). DISC1 was originally associated with schizophrenia, bipolar disorder, and major depression in a Scottish pedigree (St. Clair et al., Reference St. Clair, Blackwood, Muir, Carothers, Walker, Spowart and Evans1990) and has since been associated with schizophrenia in many studies, though findings are mixed (Brandon & Sawa, Reference Brandon and Sawa2011). DISC1 is thought to be involved in a variety of neurodevelopmental processes, including neuronal proliferation and migration as well as dendritic spine regulation and synaptic maintenance (Brandon & Sawa, Reference Brandon and Sawa2011). In rodents, fetuses exposed to maternal viral infection who carried a mutant DISC1 variant exhibited more extreme changes in pro- and anti-inflammatory cytokines in the brain compared to groups exposed to only one risk factor (Abazyan et al., Reference Abazyan, Nomura, Kannan, Ishizuka, Tamashiro, Nucifora and Pletnikov2010). Neural changes were observed in adulthood, including poorer hypothalamic–pituitary–adrenal axis reactivity and decreased serotonin neurotransmission and dendritic spine density in the hippocampus, lower amygdala volumes, and decreased lateral ventricle volume. Behaviorally, these offspring exhibited anxiety and depressive-like responses and altered social behavior. However, these behavioral deficits were only observed if the DISC1 variant was expressed across the life span. Subsequent work identified that similar effects are observed in fetuses exposed to IL-6 during the prenatal period with a variant of DISC1 that produces schizophrenia-related biological and behavioral phenotypes (Lipina, Zai, Hlousek, Roder, & Wong, Reference Lipina, Zai, Hlousek, Roder and Wong2013). Similar interactions between genetic variants and prenatal cytokine exposure on neurodevelopment and behavior have been observed for Nurr1, implicated in dopaminergic system development (Vuillermot et al., Reference Vuillermot, Joodmardi, Perlmann, Ove Ogren, Feldon and Meyer2012), and CHRNA7, a component of nicotinic acetylcholine receptors that are important for cognitive function (Wu, Adams, Stevens, Chow, & Patterson, Reference Wu, Adams, Stevens, Chow and Patterson2015).
Broadly, these findings suggest that genetic variants implicated in psychiatric illness and cytokine exposure during the prenatal period may interact in a way that potentiates prenatal exposure to pro-inflammatory cytokines and downstream effects on neurodevelopment and behavior. Though the effect of a given interaction between a genetic variant and a particular cytokine is likely small, the aggregate influence of many such interactions could be sufficient to alter neurodevelopment in ways that increase risk for the development of schizophrenia. Many of the 108 schizophrenia-associated genetic loci identified in the largest genome-wide significance study to date (Ripke et al., Reference Ripke, Neale, Corvin, Walters, Farh, Holmans and O'Donovan2014) may be preferentially expressed during middle to late fetal development (Ohi et al., Reference Ohi, Shimada, Nitta, Kihara, Okubo, Uehara and Kawasaki2016), and inflammation tends to induce changes in many cytokines at once (Saito, Nakashima, Shima, & Ito, Reference Saito, Nakashima, Shima and Ito2010). There is potential for huge numbers of these interactions to take place in response to maternal inflammation during pregnancy, but only for individuals with variants in many of these genes. This possibility may explain why most individuals exposed to infection during prenatal development or birth complications do not exhibit signs of adverse neurodevelopment, yet for certain individuals these exposures alter neurodevelopment in ways that increase risk for schizophrenia. The potentiating effect of these interactions may also explain why effect sizes tend to be small for variants of genes implicated in schizophrenia in genetic studies. Further investigation of interactions between genetic variants associated with schizophrenia and cytokine exposure on neurodevelopment may yield more insight into mechanistic pathways that can lead to schizophrenia, and could ultimately provide novel platforms for prevention.
Summary
In sum, cytokines may mediate the association between prenatal inflammation and risk for schizophrenia by affecting neurodevelopment. Elevations in maternal cytokines during pregnancy have been associated with risk for schizophrenia. Rodent models suggest these cytokines could affect the development of systems implicated in the disorder such as synaptic formation, oligodendrocyte maturation, and interneuron function. It is plausible that the composite effect of many interactions between genetic variants implicated in schizophrenia and cytokines leads to specific changes in neurodevelopment that increase risk for the disorder. Further research to assess these potential interactions could produce more precise insight into the etiology of schizophrenia.
Complement
A second candidate molecular mechanism that may link prenatal inflammation with risk for psychosis is the complement system. The complement cascade is a component of the innate immune system consisting of proteins (e.g., C1q, C3–C9, and their receptors) that “tag” and deactivate pathogens and host cellular debris for removal by phagocytic cells (Mayilyan, Weinberger, & Sim, Reference Mayilyan, Weinberger and Sim2008; Stephan et al., Reference Stephan, Barres and Stevens2012). In the CNS, complement proteins are produced by microglia and astrocytes and are massively generated in response to markers of injury or inflammation, such as C-reactive protein (Stephan et al., Reference Stephan, Barres and Stevens2012). Neurons are particularly vulnerable to damage by the complement system (Orsini et al., Reference Orsini, de Blasio, Zangari, Zanier and de Simoni2014), and uncontrolled complement activation has been implicated in neurodegenerative disorders (Orsini et al., Reference Orsini, de Blasio, Zangari, Zanier and de Simoni2014). Recent evidence suggests that complement protein expression also plays a role in typical neurodevelopmental events, such as neuronal differentiation and migration and synaptic pruning (Mayilyan et al., Reference Mayilyan, Weinberger and Sim2008; Sekar et al., Reference Sekar, Bialas, de Rivera, Davis, Hammond, Kamitaki and McCarroll2016; Stephan et al., Reference Stephan, Barres and Stevens2012). Thus, perturbations to the complement system due to exposure to maternal inflammation during pregnancy may also alter neurodevelopment in ways that increase risk for schizophrenia (Mayilyan et al., Reference Mayilyan, Weinberger and Sim2008; Stephan et al., Reference Stephan, Barres and Stevens2012).
Potential links between complement exposure and risk for schizophrenia
Complement gene expression and protein levels are altered following immune challenge with bacterial (Gewurz, Shin, & Mergenhagen, Reference Gewurz, Shin and Mergenhagen1968) and viral components (Sahu, Isaacs, Soulika, & Lambris, Reference Sahu, Isaacs, Soulika and Lambris1998). There is increasing recognition that the complement system can be activated by noninfectious antigens as well, such as hypoxic-ischemic birth complications (Cowell, Plane, & Silverstein, Reference Cowell, Plane and Silverstein2003), broad markers of inflammation such as C-reactive protein (Stephan et al., Reference Stephan, Barres and Stevens2012), and antibodies to gluten or dairy (Severance et al., Reference Severance, Alaedini, Yang, Halling, Gressitt, Stallings and Yolken2012). This pattern suggests that it is plausible that the associations observed between prenatal inflammatory insults and risk for schizophrenia may be mediated in part by complement system activation. However, there is a paucity of studies examining complement proteins during pregnancy and their association with risk for schizophrenia. One human study has identified an association between maternal C1q levels at delivery and risk for schizophrenia among offspring (Severance et al., Reference Severance, Gressitt, Buka, Cannon and Yolken2014; Figure 1). More rodent and human research is needed to assess whether levels of other complement proteins at specific time points during development are associated with risk for schizophrenia.
Complement proteins in neurodevelopment
Preliminary evidence suggests that the complement pathway is involved in typical neurodevelopment (Stephan et al., Reference Stephan, Barres and Stevens2012). The complement proteins are clearly implicated in normative experience-dependent synaptic pruning (Sekar et al., Reference Sekar, Bialas, de Rivera, Davis, Hammond, Kamitaki and McCarroll2016; Stephan et al., Reference Stephan, Barres and Stevens2012). Glial-produced C1q and C3, and increasingly C4, have been localized to immature neurons and have been shown to be critical for synaptic pruning in rodent models of retinal ganglion neuron maturation (Chu et al., Reference Chu, Jin, Parada, Pesic, Stevens, Barres and Prince2010; Sekar et al., Reference Sekar, Bialas, de Rivera, Davis, Hammond, Kamitaki and McCarroll2016; Stephan et al., Reference Stephan, Barres and Stevens2012). In contrast, the complement system does not seem to be implicated in synaptic formation (Stephan et al., Reference Stephan, Madison, Mateos, Fraser, Lovelett, Coutellier and Barres2013). Complement proteins C3 and C5 may promote the proliferation and migration of neural precursor cells in young and adult rodents in many regions of the brain, including the hippocampus and cerebellum (Bénard et al., Reference Bénard, Raoult, Vaudry, Leprince, Falluel-Morel, Gonzalez and Fontaine2008; Moriyama et al., Reference Moriyama, Fukuhara, Britschgi, He, Villeda, Molina and Holers2011; Shinjyo, Ståhlberg, Dragunow, Pekny, & Pekna, Reference Shinjyo, Ståhlberg, Dragunow, Pekny and Pekna2009). Initial evidence suggests that C1q may be associated with a subset of inhibitory interneurons, though the specific role played by C1q is unclear (Stephan et al., Reference Stephan, Madison, Mateos, Fraser, Lovelett, Coutellier and Barres2013). In addition, complement proteins have been implicated in priming helper T cells, peripheral immune cells that are similar to microglia, toward pro- or anti-inflammatory phenotypes (Yamamoto, Fara, Dasgupta, & Kemper, Reference Yamamoto, Fara, Dasgupta and Kemper2013). This set of findings suggests that the complement system is involved in a variety of neurodevelopmental processes, but the details of this involvement remain to be elucidated.
Potential influence of complement elevations on neurodevelopment of schizophrenia
The clearest potential etiological connection between the complement system and schizophrenia is via synaptic pruning (Sekar et al., Reference Sekar, Bialas, de Rivera, Davis, Hammond, Kamitaki and McCarroll2016). Most synaptic pruning takes place after birth (Figure 1), so complement elevations during the prenatal period may not influence synaptic pruning specifically. However, elevations in complement in the late second to third trimester, when glial programming may be particularly vulnerable (Bilbo & Schwarz, Reference Bilbo and Schwarz2009), could sensitize microglia to future immune challenges and have implications for future neurodevelopment. For example, reactive astrocytes respond to inflammatory markers by expressing molecules including C1q that affect synapse formation (Eroglu & Barres, Reference Eroglu and Barres2015). It is possible that sensitized microglia may release more C1q in response to inflammation, which could contribute to overpruning of synapses in childhood or adolescence (i.e., multiple hits hypothesis; Bilbo & Schwarz, Reference Bilbo and Schwarz2009; Meyer, Reference Meyer2013; Monji et al., Reference Monji, Kato and Kanba2009).
The complement system has also been associated with proliferation and migration of pyramidal neurons and interneurons (Bénard et al., Reference Bénard, Raoult, Vaudry, Leprince, Falluel-Morel, Gonzalez and Fontaine2008; Stephan et al., Reference Stephan, Madison, Mateos, Fraser, Lovelett, Coutellier and Barres2013). This suggests that elevations in complement proteins during the prenatal period may interfere with the development of these systems. However, not enough is known about the typical role of complement proteins in these processes or about exposures associated with risk for schizophrenia to make clear predictions about how this relates to the etiology of schizophrenia. Additional research identifying the role of complement proteins in typical neurodevelopment and the consequences of exposure to altered levels of these proteins in rodents, and studies examining prenatal complement exposure and risk for schizophrenia, are needed to elucidate the potential role of complement proteins in the development of schizophrenia.
Potential interactions between complement and genetic vulnerability to schizophrenia
Like cytokines, elevations in complement proteins likely contribute to a neurodevelopmental risk trajectory for schizophrenia specifically via interactions with genetic variants implicated in the disease. Investigation of potential interactions in rodent models is needed to explore this possibility. An avenue that may be fruitful is exploration of interactions between genetic variants that affect microglial function and prenatal complement exposure. Genetic variants that interfere with microglial regulation or prime microglia toward a pro-inflammatory phenotype could plausibly be associated with more microglial-mediated neural damage and/or pruning abnormalities in childhood and adolescence. Individuals with such variants who are exposed to elevated complement in the late prenatal period could undergo microglial sensitization, leading to exacerbated neural damage in response to inflammatory challenges in later childhood and adolescence. Microglial activity may be particularly important in the early development of psychosis in adolescence, as elevated microglial activation as assessed using positron emission tomography of TSPO radioligands has been observed in ultra-high risk prodromes (Bloomfield et al., Reference Bloomfield, Selvaraj, Veronese, Rizzo, Bertoldo, Owen and Howes2016) and findings with this technique are mixed in first episode (Hafizi et al., Reference Hafizi, Tseng, Rao, Selvanathan, Kenk, Bazinet and Mizrahi2017; van Berckel et al., Reference van Berckel, Bossong, Boellaard, Kloet, Schuitemaker, Caspers and Kahn2008) and chronic schizophrenia (Doorduin et al., Reference Doorduin, de Vries, Willemsen, de Groot, Dierckx and Klein2009; Kenk et al., Reference Kenk, Selvanathan, Rao, Suridjan, Rusjan, Remington and Mizrahi2015). It is possible that individuals with particularly high microglial activity in adolescence in addition to another vulnerability factor, such as NMDA receptor hypofunction, could be at even higher risk of developing psychosis. NMDA receptor hypofunction, resulting in reduced transmission of excitatory glutamate into the postsynaptic neuron, has long been thought to be a key component of the pathophysiology of schizophrenia (Olney, Newcomer, & Farber, Reference Olney, Newcomer and Farber1999). NMDA receptor activity at synapses is thought to be critical to the growth and stabilization of dendritic spines (Arnsten, Reference Arnsten2011), and spines that are stable are thought to be less vulnerable to being pruned (Glausier & Lewis, Reference Glausier and Lewis2013). It is possible that individuals who have less effective NMDA receptors have greater numbers of weak spines, which could be less likely to survive normative synaptic reshaping during the first few years of life. Individuals with overactive microglia and inappropriately weak synaptic connections may be particularly likely to experience abnormally high rates of synaptic pruning in adolescence. These individuals may be at particularly high risk to cross below a threshold of synaptic density needed to support coordinated neuronal firing, resulting in poorer cognitive function (Homayoun & Moghaddam, Reference Homayoun and Moghaddam2007) and potentially the onset of psychosis (Cannon, Reference Cannon2015; Feinberg, Reference Feinberg1982).
In all, there are many possible interactions between variants affecting microglial priming and regulation, synaptic strength and functioning, and prenatal complement system activity that could be involved in the etiology of schizophrenia. Further work assessing interactions such as these would allow us to test theories linking the complement system with synaptic pruning in schizophrenia and further our understanding of the role that the complement system may play in the etiology of schizophrenia.
Summary
Initial evidence suggests that complement proteins may mediate the association between prenatal inflammation and risk for schizophrenia by affecting neurodevelopment. The complement system is most clearly implicated in synaptic pruning, but may play a role in neuronal growth and migration and glial priming as well. Interactions between complement proteins and genetic variants in these systems may contribute to specific patterns observed in schizophrenia, such as faster rates of synaptic pruning in adolescence. Further research is needed to clarify the role of complement proteins in neurodevelopmental processes, identify periods of development and specific proteins that are associated with schizophrenia risk, and assess interactions with other vulnerability markers to establish how complement proteins may play a role in the etiology of schizophrenia.
Implications for the Development of Schizophrenia
Disruption of neurodevelopment by synergistic interactions between prenatal cytokine and complement exposure and genetic risk for schizophrenia ought to become apparent as neural systems emerge and are refined across postnatal development. The nature of the neurodevelopmental systems affected and the age at which deficits emerge may yield more insight into different etiological pathways to schizophrenia and the mechanisms by which prenatal cytokine and complement exposure may contribute to increasing risk. On average, individuals who develop schizophrenia exhibit markers of neurodevelopmental abnormalities in childhood, such as poorer verbal memory and perceptual–motor performance (Bearden et al., Reference Bearden, Rosso, Sanchez, Hadley, Nuechterlein and Cannon2003; Brown et al., Reference Brown, Cohen, Harkavy-Friedman, Babulas, Malaspina, Gorman and Susser2001; Cannon et al., Reference Cannon, Bearden, Hollister, Rosso, Sanchez and Hadley2000; Ellman et al., Reference Ellman, Yolken, Buka, Torrey and Cannon2009), motor abnormalities (e.g., atypical postures and movements; Cannon et al., Reference Cannon, Rosso, Bearden, Sanchez and Hadley1999), and abnormal behavioral patterns (Bearden et al., Reference Bearden, Rosso, Houister, Sanchez, Hadley and Cannon2000). However, individuals with early-onset psychosis tend to exhibit widespread and significant cognitive deficits early in childhood, whereas those who develop psychosis later in adolescence or adulthood tend to exhibit more focal and less severe cognitive deficits (Rajji, Ismail, & Mulsant, Reference Rajji, Ismail and Mulsant2009). Some evidence suggests that cognitive deficits in childhood may be more profound in individuals exposed to prenatal inflammatory challenges who later develop schizophrenia compared to cases who did not experience prenatal insults (Brown et al., Reference Brown, Cohen, Harkavy-Friedman, Babulas, Malaspina, Gorman and Susser2001, Reference Brown, Vinogradov, Kremen, Poole, Deicken, Penner and Schaefer2009; Ellman et al., Reference Ellman, Yolken, Buka, Torrey and Cannon2009), though evidence is mixed (Bearden et al., Reference Bearden, Rosso, Houister, Sanchez, Hadley and Cannon2000; Cannon et al., Reference Cannon, Bearden, Hollister, Rosso, Sanchez and Hadley2000). It is possible that exposure to prenatal inflammation sets all vulnerable individuals on a more severe developmental trajectory that leads to cognitive deficits earlier in development than they would have otherwise experienced. This pattern would suggest that prenatal inflammation may be associated with broad, nonspecific insults to neurodevelopment that emerge relatively early in childhood, perhaps such as myelination or synaptic refinement across the prefrontal cortex. Another possibility is that prenatal inflammation affects neurodevelopment for individuals who are specifically predisposed to risk trajectories that tend to emerge in childhood and has a minimal effect on individuals who are predisposed to trajectories that tend to emerge during adolescence. This hypothesis could suggest more specific interactions between inflammatory factors and genetic factors that affect regions involved in cognition in childhood. It is also possible that prenatal inflammation can interact with genetic variants and have effects that are not “unmasked” until adolescent developmental processes take place, such as synaptic refinement (Weinberger, Reference Weinberger1987). Another such example could be interactions with the endocannabinoid system. Expression levels of the endocannabinoid receptor CB1R follow an inverted U-shaped curve that peaks in adolescence in layer III of the monkey dorsolateral prefrontal cortex (Keshavan, Giedd, Lau, Lewis, & Paus, Reference Keshavan, Giedd, Lau, Lewis and Paus2014). Activation of CB1Rs suppresses GABA release from interneurons that fire on excitatory neurons, and overactivation of these receptors is thought to disrupt typical development of interneuron–pyramidal neuron circuitry in adolescence and increase risk for psychosis (Keshavan et al., Reference Keshavan, Giedd, Lau, Lewis and Paus2014). Rodents exposed to viral components during pregnancy followed longitudinally exhibited decreased CB1R density in the globus pallidus in adolescence and increased density in the hypothalamus and sensory cortex in adulthood compared to controls (Verdurand et al., Reference Verdurand, Dalton, Nguyen, Grégoire, Zahra, Wyatt and Zavitsanou2014). This set of findings suggests that prenatal exposure to inflammation may have effects on the endocannabinoid system that do not become apparent until extreme periods of receptor density such as adolescence. In combination with other risk factors such as cannabis use during adolescence, prenatal inflammation could increase risk for adolescent-onset psychosis as well.
It is likely that each of these possibilities is represented in many different combinations of genetic variants and prenatal cytokine and complement elevations. Monitoring cognitive and behavioral development among those at high genetic risk for schizophrenia who are exposed to prenatal inflammation could help elucidate these potential pathways to the disorder, and follow-up rodent studies could be used to assess possible mechanisms. Ultimately, this knowledge could help distinguish between different etiological pathways to schizophrenia and provide novel opportunities for treatment and intervention.
Potential for neonatal interventions
Ultimately, a more specific understanding of cytokine and complement system interactions could help identify potential interventions following prenatal complications that could help prevent a developmental risk trajectory for schizophrenia. Though we do not yet have sufficient knowledge to implement interventions in human neonates, rodent studies suggest that anti-inflammatory interventions at birth may be able to prevent neurodevelopmental damage typically observed following prenatal inflammatory exposure. For instance, elevated levels of anti-inflammatory proteins following prenatal inflammatory insults may counteract the deleterious effects of elevations in pro-inflammatory proteins on neurodevelopment. In a rodent model, administration of IL-1RA after birth eliminated deficits in behavior typically observed following bacterial infection, hypoxic-ischemic injury, or both (Girard et al., Reference Girard, Sébire, Brochu, Briota, Sarret and Sébire2012). Similarly, elevations in IL-10 have been shown to mitigate the effects of prenatal bacterial infection on white matter damage (Rodts-Palenik et al., Reference Rodts-Palenik, Wyatt-Ashmead, Pang, Thigpen, Cai, Rhodes and Bennett2004) and of prenatal viral infection on elevations in pro-inflammatory cytokines and deficits in exploratory behavior, sensorimotor gating, and inhibitory learning (Meyer et al., Reference Meyer, Murray, Urwyler, Yee, Schedlowski and Feldon2008). However, elevations in prenatal IL-10 in the absence of a pro-inflammatory challenge were associated with behavioral deficits linked to viral infection, including reduced exploratory behavior and inhibitory learning (Meyer et al., Reference Meyer, Murray, Urwyler, Yee, Schedlowski and Feldon2008). These findings suggest that administration of anti-inflammatory cytokines may be harmful rather than beneficial if it occurs in the absence of a pro-inflammatory challenge. In addition, specific cytokines (e.g., TNFα and GM-CSF) have been found to promote complement-mediated phagocytosis of antigens by peripheral macrophages (Collins & Bancroft, Reference Collins and Bancroft1992). This result suggests that targeting certain cytokines could have effects on complement-mediated influences on neurodevelopment. Thus, it is plausible that anti-inflammatory treatment in the early postnatal period could be used to counteract excessive inflammation in the developing brain to support typical neurodevelopment.
The possibility of early life interventions to support healthy neurodevelopment and decrease risk for schizophrenia is very encouraging and has the potential to become a novel platform for prevention. Before such interventions can be developed and ethically implemented, much more knowledge is needed to ensure that treatment will counteract rather than exacerbate immune imbalances in the fetal brain. Domains of knowledge that need to be expanded to support this end goal are discussed, including typical and atypical immune signaling patterns during pregnancy, immune patterns associated with particular prenatal exposures, the potential for synergy between prenatal immune exposures, correspondence between maternal and fetal immune protein levels, studying immune proteins in isolation versus in signaling pathways, and interactions with other biological systems.
Advances needed to support neonatal intervention
Typical immune signaling during pregnancy and risk patterns
There is some indication that the maternal immune system changes throughout pregnancy, likely to promote tolerance of fetal tissue (Girardi, Bulla, Salmon, & Tedesco, Reference Girardi, Bulla, Salmon and Tedesco2006). For example, the first trimester of pregnancy is characterized by high levels of anti-inflammatory IFNγ levels and low levels of typically pro-inflammatory IL-6 in maternal serum, whereas by the third trimester of pregnancy IL-6 levels are high and IFNγ levels are low (Aris, Lambert, Bessette, & Moutquin, Reference Aris, Lambert, Bessette and Moutquin2008). In contrast, C3, C4, and C5 levels are higher in the serum of pregnant women compared to nonpregnant women, and remain at consistent levels in maternal serum throughout pregnancy (Richani et al., Reference Richani, Soto, Romero, Espinoza, Chaiworapongsa, Nien and Mazor2005). These findings suggest that immune pathway activity may change across pregnancy, with important implications for maternal and fetal reactivity to immune challenges. However, only a few studies have assessed maternal cytokine levels longitudinally (Holtan et al., Reference Holtan, Chen, Kaimal, Creedon, Enninga, Nevala and Markovic2015; Ross et al., Reference Ross, Miller, Culhane, Grobman, Simhan, Wadhwa and Borders2016), and typical patterns of marker levels across pregnancy have yet to be established. Characterizing the typical patterns of cytokines and complement in maternal and fetal serum will help clarify which alterations in cytokine and complement levels are atypical for that period in development, and could yield insight into why similar prenatal exposures can have different effects at different times. For instance, preterm-equivalent rodents exposed to bacteria and hypoxic-ischemic injury at birth exhibited upregulation of only anti-inflammatory cytokines, whereas term-equivalent rodents exposed to these two insults exhibited upregulation of both pro-inflammatory and anti-inflammatory cytokines (Brochu, Girard, Lavoie, & Sébire, Reference Brochu, Girard, Lavoie and Sébire2011). This knowledge may also nominate periods of development and cellular systems that may be particularly associated with risk for schizophrenia. Thus far, the studies that have examined whether elevations in maternal cytokines during pregnancy are associated with risk for schizophrenia in human samples have been cross-sectional (Allswede et al., Reference Allswede, Buka, Yolken, Torrey and Cannon2016; Brown, Hooton, et al., Reference Brown, Hooton, Schaefer, Zhang, Petkova, Babulas and Susser2004; Buka, Tsuang, Torrey, Klebanoff, Bernstein, et al., Reference Buka, Tsuang, Torrey, Klebanoff, Bernstein and Yolken2001; Ellman et al., Reference Ellman, Deicken, Vinogradov, Kremen, Poole, Kern and Brown2010; Goldstein et al., Reference Goldstein, Cherkerzian, Seidman, Donatelli, Remington, Tsuang and Buka2014). Elucidating different prenatal inflammatory trajectories and examining their association with postnatal development may further specify different etiological pathways that can increase risk for schizophrenia.
Inflammatory patterns associated with specific exposures
Prenatal immune activation increases fetal inflammation broadly, but certain exposures may be more strongly associated with specific markers. Generally, injections of bacterial and viral components are known to induce elevations in IL-1β, IL-6, and TNFα in maternal serum. Viral components may also particularly activate IFNγ (Bilbo et al., Reference Bilbo, Biedenkapp, Der-Avakian, Watkins, Rudy and Maier2005; Boksa, Reference Boksa2010; Cai et al., Reference Cai, Pan, Pang, Evans and Rhodes2000; Smith et al., Reference Smith, Li, Garbett, Mirnics and Patterson2007). Elevations in IL-1β and IL-2 have been observed following hypoxic-ischemic injury (Girard, Kadhim, et al., Reference Girard, Kadhim, Larouche, Roy, Gobeil and Sébire2008; Girard, Larouche, et al., Reference Girard, Larouche, Kadhim, Rola-Pleszczynski, Gobeil and Sébire2008). Nevertheless, there is some evidence suggesting that distinct cytokines have been implicated in mediating the effects of maternal inflammation on neurodevelopment. The effects of viruses on neurodevelopment may be mediated by changes in IL-6 specifically, and not IL-1β or IFNγ (Smith et al., Reference Smith, Li, Garbett, Mirnics and Patterson2007). In contrast, IL-1β may be particularly involved in neurodevelopmental changes resulting from bacteria (Girard, Tremblay, Lepage, & Sébire, Reference Girard, Tremblay, Lepage and Sébire2010). Assessing cognitive changes in response to viral and bacterial infection similarly suggests many common deficits and the potential for distinct deficits. Exposure to each pathogen has been shown to lead to poorer prepulse inhibition, spatial learning, and novel object recognition. Latent inhibition may be particularly disrupted following viral infection (Boksa, Reference Boksa2010). Further work in both rodent models and longitudinal human samples with birth record data is needed to assess whether certain prenatal exposures are consistently associated with specific cytokines and neurodevelopmental deficits or if the effects are consistent across exposures. There may be less specificity to exposure type in the complement system, as C3 seems to be activated by bacteria, viruses, and hypoxic-ischemic injury (Cowell et al., Reference Cowell, Plane and Silverstein2003; Gewurz et al., Reference Gewurz, Shin and Mergenhagen1968; Sahu et al., Reference Sahu, Isaacs, Soulika and Lambris1998). However, more work is needed to assess whether certain exposures are associated with distinct patterns of complement system activity and neurodevelopmental deficits as well. If there is specificity associated with particular exposures, this information could eventually be used to develop treatments for neonates designed to counteract the specific associated inflammatory patterns. If exposures exhibit the same general pattern, a common treatment could be used to support optimal neurodevelopment for all neonates.
Potential for synergism with multiple inflammatory insults
As similar cytokines and complement proteins tend to be secreted in response to multiple types of prenatal insults, exposure to multiple prenatal inflammatory events may lead to a potentiated inflammatory effect that puts individuals at greater risk for altered neurodevelopment. For example, rats exposed to either bacterial infection in the last few days of gestation or hypoxic-ischemic insult at birth exhibited an increase in proinflammatory IL-1β and IL-2 levels and a decrease in anti-inflammatory IL-1RA levels in the brain in the first 2 postnatal days, and rats exposed to both exhibited the greatest changes in these levels (Girard, Kadhim, et al., Reference Girard, Kadhim, Larouche, Roy, Gobeil and Sébire2008; Girard, Larouche, et al., Reference Girard, Larouche, Kadhim, Rola-Pleszczynski, Gobeil and Sébire2008). In adulthood, rodents exposed to both prenatal insults exhibited the greatest changes in behavior relative to controls, such as reduced exploratory behavior (Girard et al., Reference Girard, Sébire, Brochu, Briota, Sarret and Sébire2012). Similarly, rats exposed to viral infection before hypoxic-ischemic insult at birth exhibited greater reductions in myelin basic protein in the neonatal period compared to rodents exposed only to hypoxia-ischemia (Stridh et al., Reference Stridh, Mottahedin, Johansson, Valdez, Northington, Wang and Mallard2013). Higher levels of pro-inflammatory cytokines and lower levels of anti-inflammatory microglia were observed following viral infection and prior to hypoxic-ischemic insult, and could explain the stronger effect of exposure to viral infection on neural damage (Stridh et al., Reference Stridh, Mottahedin, Johansson, Valdez, Northington, Wang and Mallard2013). However, similar studies have identified that infection prior to hypoxia-ischemia can have a preconditioning effect that mitigates damage to the developing brain (Mallard & Hagberg, Reference Mallard and Hagberg2007). This contradiction could be explained by differences in length of time between infection and hypoxic-ischemic injury. As typical inflammatory responses first exhibit an increase in pro-inflammatory factors and then an increase in anti-inflammatory factors, different lengths of time between the first and second insult could result in different environmental contexts when the second insult hits, yielding different effects on overall inflammation (Mallard & Hagberg, Reference Mallard and Hagberg2007). A more detailed understanding of these factors could help us identify infants who are at particular risk for cytokine-mediated neurodevelopmental damage and potentially inform the strength of anti-inflammatory treatment needed.
Correspondence between maternal and fetal immune signaling
Examining the association between maternal cytokine and complement levels and risk for schizophrenia among offspring is a critical step toward translation of rodent literature to humans. However, these studies are vulnerable to the assumption that elevations in maternal serum reflect changes in the fetal brain. Rodents studies suggest that changes in maternal cytokine levels in serum are sometimes, but not always, consistent with cytokine changes in the placenta, fetal serum, and fetal brain (Ashdown et al., Reference Ashdown, Dumont, Ng, Poole, Boksa and Luheshi2006; Garay, Hsiao, Patterson, & McAllister, Reference Garay, Hsiao, Patterson and McAllister2013; Gilmore, Jarskog, & Vadlamudi, Reference Gilmore, Jarskog and Vadlamudi2005; Urakubo et al., Reference Urakubo, Jarskog, Lieberman and Gilmore2001). Apparent inconsistencies may be influenced by the timing of measurement of cytokine levels. For example, injection of bacterial components has been shown to cause a rapid increase in IL-1β (~1 hr) and a more gradual and protracted increase in TNFα (~4–24 hr) in the fetal brain (Cai et al., Reference Cai, Pan, Pang, Evans and Rhodes2000). This suggests that some elevations in cytokines in the fetal brain may be missed if only one measurement is taken. True differences between maternal and fetal cytokine levels may be influenced by the transfer of the inflammatory response at the placenta. Cytokines are thought to not be able to cross from maternal to fetal tissue, with the possible exception of IL-6 (Meyer et al., Reference Meyer, Feldon and Yee2009). The maternal inflammatory response at the boundary of the placenta is thought to induce an inflammatory response from the fetal side of the placenta, much like the transduction of cytokines at the blood–brain barrier (Meyer et al., Reference Meyer, Feldon and Yee2009). Thus, it is possible that changes in cytokine signaling are made at these interfaces. Less is known about the consistency of complement signaling in maternal and fetal serum and tissues. Studies of the maternal–fetal interface suggest that complement inhibitors are highly expressed to prevent potential damage of placental tissue (Girardi et al., Reference Girardi, Bulla, Salmon and Tedesco2006). Additional rodent studies that examine the consistency of cytokine and complement expression in maternal serum and the fetal brain across multiple time points could help clarify which cytokines are transduced from maternal to fetal tissue and which are not. Further investigation of roles of specific immune proteins in neurodevelopment, and comparison of these findings to changes observed (e.g., neural structure) in offspring whose mothers were exposed to that protein during neurodevelopment, could also help assess whether the changes observed in offspring match those that would be expected. Once sufficient rodent literature has been established to generate predicted effects of changes to specific maternal immune proteins on fetal development, these predictions could be assessed in human samples utilizing maternal serum collected during pregnancy and neonatal imaging measures.
Assessing cytokine pathways
Though previous investigations of maternal cytokine levels in association with risk for psychosis have predominantly focused on single immune markers, assessing clusters of cytokines may more comprehensively reflect the dynamics of cytokine signaling pathways (Saito et al., Reference Saito, Nakashima, Shima and Ito2010). One such pathway could group cytokines based on their association with pathways of immune cell activation in peripheral blood mononuclear cells: pro-inflammatory M1 macrophage polarization, which in turn activates pro-inflammatory helper T (Th) cell types Th1 and Th17 subtypes (cytokines: TNFα, IFNγ, GM-CSF, IL-1β, IL-2, IL-6, IL-12, and IL-17), and anti-inflammatory M2 macrophage polarization, which in turn activates anti-inflammatory Th2 and Treg cell types (CSF1, IL-4, IL-5, IL-10, and IL-13; Italiani & Boraschi, Reference Italiani and Boraschi2014; Ponomarev et al., Reference Ponomarev, Veremeyko and Weiner2013). Grouping cytokines by their common pathways may improve signal-to-noise ratio as well as facilitate interpretation of findings within the biological context. However, it is also possible that examining trajectories of single cytokines accounts for more signal in association with neurodevelopmental outcomes and may be most informative. Comparing groupings of cytokines versus individual assessment would help clarify which approach may be most beneficial in investigations of prenatal immune activation and neurodevelopmental outcomes, such as risk for schizophrenia.
Interactions between immune proteins and other biological mechanisms
Neurodevelopmental changes observed in response to maternal immune activation are not the result of cytokines and complement proteins alone. Cytokines are known to interact with a variety of other factors that influence neurodevelopment. For example, IL-1β has been shown to interfere with the neuroprotective effects of brain-derived neurotrophic factor in cultured neurons (Tong, Balazs, Soiampornkul, Thangnipon, & Cotman, Reference Tong, Balazs, Soiampornkul, Thangnipon and Cotman2008). Cytokines are known to interact with the hypothalamic–pituitary–adrenal axis as well, which regulates stress reactivity. For example, IL-1β and IL-6 can induce upregulation of corticotropin-releasing hormone and vice versa, leading to increased activation of both systems (Theoharides, Weinkauf, & Conti, Reference Theoharides, Weinkauf and Conti2004). Cytokines also increase oxidative stress through activation of the tryptophan catabolite pathway (Anderson et al., Reference Anderson, Berk, Dodd, Bechter, Altamura, Dell'Osso and Maes2013). Similarly, complement proteins may interact with other neurodevelopmental factors. Elevations in C3a in pituitary cultures has been shown to increase release of growth hormone, prolactin, and adrenocorticotropic hormone (Francis et al., Reference Francis, Lewis, Akatsu, Monk, Cain, Scanlon and Gasque2003). Oxidative stress is known to activate the complement system as well (Collard et al., Reference Collard, Vä, Morrissey, Agah, Rollins, Reenstra and Stahl2000). This suggests that neurodevelopmental changes observed in response to prenatal immune challenge are the result of interactions between these pathways. More work is needed to elucidate potential interactions between these biological systems.
Conclusion
The association between maternal immune activation during pregnancy and risk for schizophrenia may be mediated in part by the influence of cytokines and complement proteins on neurodevelopment. Alterations in levels of these proteins in the prenatal period may interact with genetic deficits implicated in schizophrenia to perturb neurodevelopmental processes such as synaptic formation and pruning, interneuron proliferation and migration, and myelination. These cellular changes could contribute to structural and network deficits that affect cognition and behavior across development, ultimately leading to schizophrenia. A more thorough understanding of the ways in which prenatal cytokines and complement may influence subsequent development may inform our understanding of different etiological pathways to schizophrenia, though much more work is warranted. Ultimately, this knowledge could be used to create anti-inflammatory treatments to support healthy postnatal neurodevelopment, and could provide a novel platform of intervention to prevent the development of schizophrenia in at-risk individuals.

