


The desert-dwelling Western Spadefoot toad lays her eggs in pools of rainwater during the Spring. As the desert heats up, the tadpoles confront the stress of rapidly evaporating water that shrinks their life support system. Detection by the tadpole of this life-threatening stress results in the synthesis and release of corticotropic-releasing hormone (CRH) into the median eminence of the hypothalamus precipitating accelerated metamorphosis, which increases the probability of survival (Denver Reference Denver1997, Reference Denver1999). If the CRH response is blocked during environmental desiccation, the rate of development is arrested and the tadpole's survival is compromised. There are long-term consequences for the surviving toad exposed to this stress because it is small and is at a disadvantage against normally developing toads in foraging for food and reproducing. The CRH response in the tadpole contributes to the trade-offs in the trilogy of adaptation. First, the CRH “response” ensures survival. Second, growth of the surviving toad is compromised. Third, reproductive competence is threatened. The questions addressed in this paper thus are as follows; ”What is CRH?” “Where does it come from?” and “Does it have anything to do with human behavior and more directly with development?”
Hypothalamic CRH: Is the Brain an Endocrine Organ?
In a prophetic monograph, Geoffrey Harris (Reference Harris1955) argued against all known facts and closely held beliefs, that the brain had endocrine properties. At the time of the Harris monograph, it was generally known that the pituitary gland had three distinguishing lobes (anterior, intermediate, and posterior) and that each lobe synthesized and released a unique profile of hormones with specific functions. The pituitary gland was known to be connected to the base of the brain by a short stalk, but direct neural control between the hypothalamus and the pituitary had been ruled out by careful studies showing that the pituitary remained functional after all possible connections had been severed (Harris, Reference Harris1937, Reference Harris1950). Harris (Harris, Mnabe, & Ruf, Reference Harris, Manabe and Ruf1969) and others (McCann & Friedman, Reference McCann and Friedman1960; McCann & Fruit, Reference McCann and Fruit1957; Krulich, Dhariwal, & McCann, Reference Krulich, Dhariwal and McCann1968; Krulich & McCann, Reference Krulich and McCann1966) had in vitro physiological evidence that neural tissue from the hypothalamus could stimulate pituitary activity. To explain this, Harris proposed that the hypothalamus synthesized hormones that were released into the portal system (capillary beds connecting the hypothalamus to the anterior pituitary) stimulating the pituitary to syntheize and release hormones into the circulation. This evidence was discounted for several reasons including the possibility that the neural tissue was impure and may have contained fragments of the pituitary. Harris had shown that cutting the portal vessels between the hypothalamus and pituitary did block the production of pituitary hormones. However, despite a distinguished career, Harris never was able to prove the existence of hypothalamic hormones.
A biochemist (Andrew Schally) and a physiologist (Roger Guillemin), inspired by Harris, working together and then apart began a highly competitive search for the structure of hypothalamic factors that would stimulate the pituitary to synthesize and release hormones (Wade, Reference Wade1981). It is laboratory lore that the initial target was a molecule that liberated adrenocorticotropic hormone (ACTH). ACTH was an important target system because the classical studies of Hans Selye (Reference Selye1950) had shown it stimulated the adrenal gland to produce hormones in response to stress. These adrenal hormones provided the metabolic basis for the general adaptation syndrome that Selye proposed as a mechanism for the organism both to cope with environmental challenge and then to succumb if the challenge became chronic. Despite many years of meticulous research, the structure of the imagined corticotropic hypothalamic hormone (CRH: Schally's group) or factor (CRF: Guillemin's group) eluded them. However, these intrepid researchers, enduring criticism from journals and funding agencies, made major discoveries in their failed search for the structure of corticotropic-releasing hormone (CRH).
Because the concentration of the hypothalamic hormones proved to be small and because of the poor sensitivity of the available assays, both research groups were required to gather hundreds of thousands of ovine (Guillemin) or porcine (Schally) hypothalamic tissue from which they initially identified three hypothalamic factors, thyrotropin-releasing hormone (TRH; Boler, Enzmann, Folkers, Bowers, & Schally, Reference Boler, Enzmann, Folkers, Bowers and Schally1969; Burgus, Dunn, Desiderio, & Guillemin, Reference Burgus, Dunn, Desiderio and Guillemin1969), luteninizing hormone releasing hormone (Schally & Bowers, Reference Schally and Bowers1964) and somatostatin (Burgus, Ling, Butcher, & Guillemin, Reference Burgus, Ling, Butcher and Guillemin1973). All three releasing hormones have a relatively simple structure ranging from 3 (TRH) to 14 (somatostatin) amino acids but have powerful influences on critical life functions. In a pattern that is common to releasing hormones (see Figure 1), TRH travels down the hypophyseal portal system to the median eminence where it collects before acting on the anterior pituitary to release thyroid stimulating hormone and prolactin. Luteninizing hormone releasing hormone stimulates the release of both follicle-stimulating hormone and luteninizing hormone, and somatostatin inhibits the anterior pituitary secretion of growth hormone and thyroid stimulating hormone. For these discoveries, Schally and Guillemin shared the Nobel Prize for Medicine in 1977.
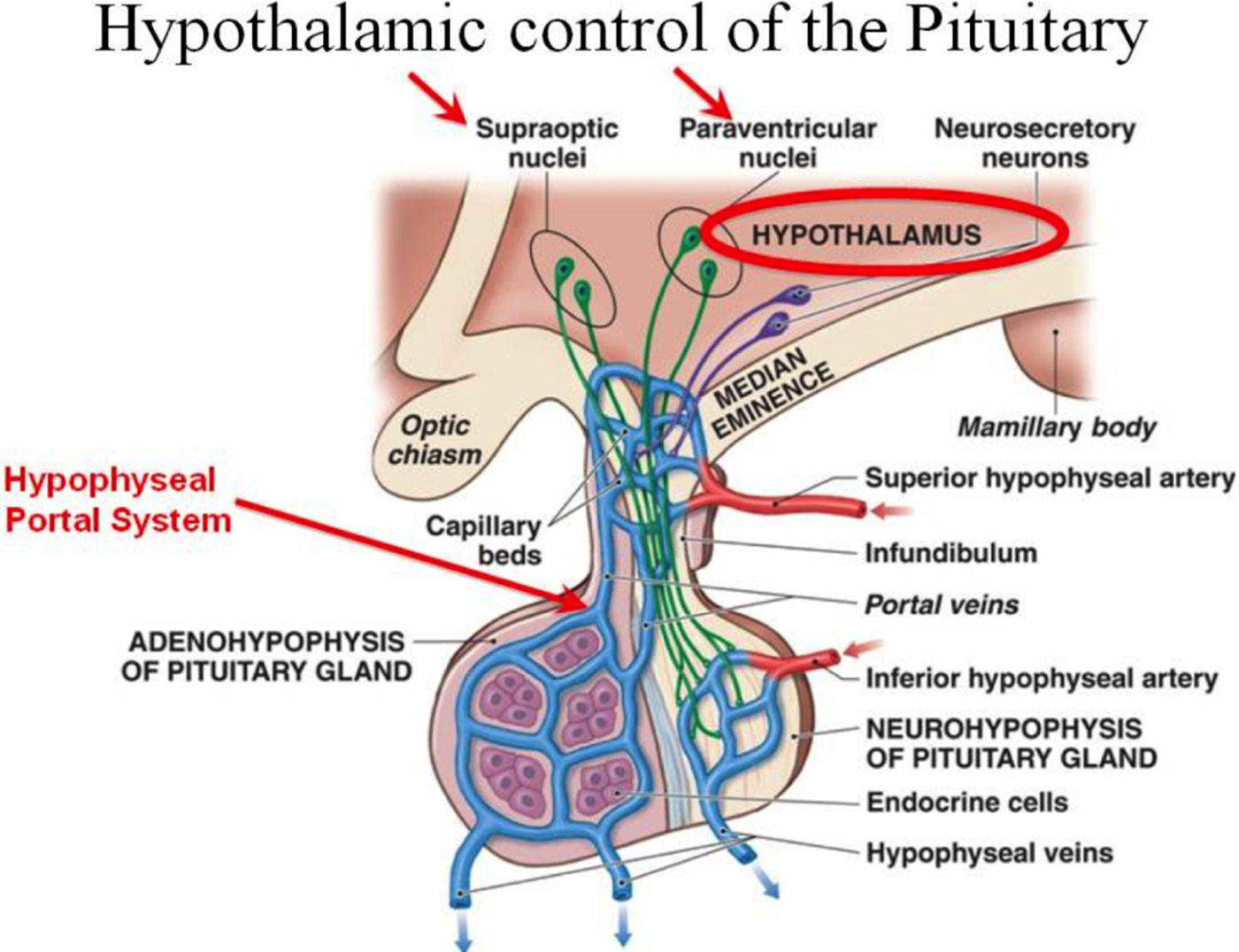
Figure 1. Releasing and inhibiting hormones are synthesized in hypothalamic neurons and collect in the median eminence (part of the hypothalamus) before entering the portal system. The portal system, in the pituitary stalk (or infundibulum), is the communication route between the hypothalamus and the pituitary gland. The hormones are released into a capillary network and transported to the pituitary.
In 1981, a team led by Wylie Vale (an earlier collaborator of Guillemin) modestly announced the sequence for CRH (reported as CRF): “We report here the purification, sequence analysis, and total synthesis of a 41-residue peptide that stimulates the secretion of corticotropin-like and 3-endorphin-like immunoactivities in vitro and in vivo” (Vale, Spiess, Rivier, & Rivier Reference Vale, Spiess, Rivier and Rivier1981, p. 1394; Figure 2). Vale's group determined CRH activity from nearly half a million fragments of ovine hypothalamus. The evidence for stimulation of both ACTH and endorphin immunoactivity was important because these two peptides reside in the anterior pituitary (along with B-LPH and several MSH fragments) within a 31K dalton, bioinactive prohormone, proopiomelanocortin (POMC; Mains & Eipper, Reference Mains and Eipper1979). Enzymes act at “weak” links on prohormones to liberate the bioactive fragments. The discovery of prohormones that contain bioactive peptides is a stable principle that pertains to peptide hormones including CRH. Specifically, the prohormone for CRH (proCRH) is a 172-residue, small proteinlike molecule that is enzymatically cleaved at the Arg152-Arg153 sequence resulting in the release of the bioactive 41-residue peptide (Brar, Sanderson, Wang, & Lowry, Reference Brar, Sanderson, Wang and Lowry1997).
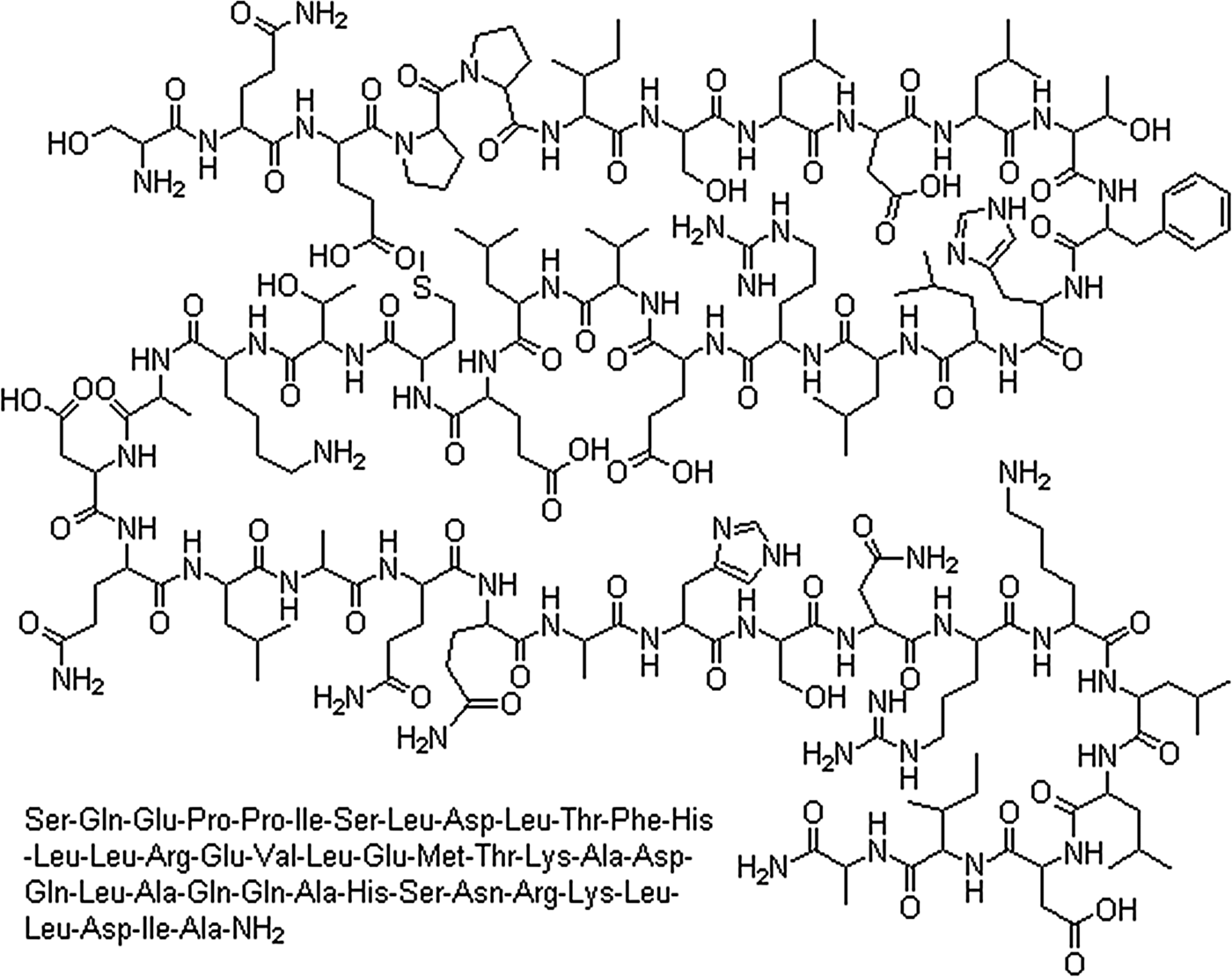
Figure 2. The structure and amino acid sequence of CRH as discovered by Vale et al. (Reference Vale, Spiess, Rivier and Rivier1981).
The gene coding for CRH (located on the long arm of chromosome 8) and the gene expression patterns of CRH are conserved among vertebrates. The expressed gene product, bioactive CRH, is distributed heterogeneously throughout the central nervous system but is localized within the neurons of the parvocellular region of the paraventricular nucleus of the hypothalamus. CRH-like peptides also are expressed in the cortex, structures of the limbic system (especially the hippocampus and amygdala), and brainstem nuclei as well as peripheral sites including the placenta as described below (Sanderson, Woods, Kemp, & Lowry, Reference Sanderson, Woods, Kemp and Lowry2000).
There were collateral consequences associated with the excitement generated by the search and discovery of the hypothalamic hormones. One major consequence was the discovery of prohormones discussed above. A second major consequence was the understanding that the labels given to peptides (and neuropeptides) often were misleading or at least limiting. Peptide hormones were labeled according to the first or most obvious function they served. One example both of the importance of recognizing the role of prohormones and of mislabeling of peptides by function was the status of melanocyte-stimulating hormone (MSH). MSH received its moniker because its synthesis and release first was observed to result in dispersion of melanophores in amphibians to darken the skin to blend with the environment. (This process is reversed by a hypothalamic inhibiting factor; MIF). However, subsequent research discovered that (a) the structure of MSH was embedded in POMC (as was ACTH and B-endorphin), (b) a bioactive core of MSH shared an amino acid sequence with ACTH, and (c) MSH had extrapigmentary neurobehavioral properties in mammals (Kastin, Plotnikoff, Schally, & Sandman, Reference Kastin, Plotnikoff, Schally, Sandman, Ehrenpreis and Kopin1976; Sandman et al., Reference Sandman, Denman, Miller, Knott, Kastin and Schally1971; Sandman, Kastin, & Schally, Reference Sandman, Kastin and Schally1969; Sandman, Miller, Kastin, & Schally, Reference Sandman, Miller, Kastin and Schally1972). The “mislabeling” is dramatic for MSH, but the presumption of limited (but significant) function for CRH also is inaccurate: CRH has functions well beyond initiating the synthesis of POMC in the anterior pituitary.
Placental CRH: Is the Placenta an Endocrine Organ?
The maternal endocrine system is profoundly altered during human pregnancy. The pituitary gland doubles in size and the output of pituitary peptides increases several fold as gestation progresses. However, it is the growth and development of a new organ, the placenta (primarily a fetal organ), in primates that is mainly responsible for the profound changes in the endocrine system including hormones of the stress circuit. Historically, the placenta was considered to be a passive organ principally responsible for filtering toxins and delivering nutrients to the fetus (Voltolini & Petraglia, Reference Voltolini, Petraglia, Fliers, Korbonits and Romijn2014). This idea has radically changed, and now the placenta is known to be a transient endocrine organ that is a major regulator of maternal and fetal physiology. A significant contribution to the appreciation of this shift in understanding the placenta is the dramatic elevations of CRH in maternal plasma during the course of human pregnancy (Campbell et al., Reference Campbell, Linton, Wolfe, Scraggs, Jones and Lowry1987; Chan et al., Reference Chan, Smith, Lewin, Brinsmead, Zhang, Cubis and Hurt1993; Goland, Wardlaw, Stark, Brown, & Frantz, Reference Goland, Wardlaw, Stark, Brown and Frantz1986; Sasaki et al., Reference Sasaki, Shinkawa, Margioris, Liotta, Sato, Murakami and Yoshinaga1987; Wolfe et al., Reference Wolfe, Patel, Campbell, Anderson, Dornhorst, Lowry and Jones1988) where CRH reaches levels observed only in the hypothalamic portal system during physiological stress (Lowry, Reference Lowry1993). Because CRH of hypothalamic origin is diluted and rapidly degraded after leaving the portal system (King, Smith, & Nicholson, Reference King, Smith and Nicholson2001), it is not detectable in the circulation, even after extreme stress, and because the concentrations of CRH in maternal plasma recede within 24 hr after delivery, it was speculated that the placenta was the source (Grino, Chrousos, & Margioris, Reference Grino, Chrusos and Margioris1987). Preliminary findings reported that a “CRF-like activity” could be detected in human placental tissues obtained at full term from spontaneous deliveries (Shibasaki, Odagiri, Shizume, & Ling, Reference Shibasaki, Odagiri, Shizume and Ling1982). Subsequent studies collected from placentae obtained from women undergoing “effective” cesarean full-term delivery confirmed that placental CRH is structurally and biochemically identical to hypothalamic CRH (Sasaki et al., Reference Sasaki, Tempst, Lotta, Margioris, Hood, Kent and Krieger1988).
Although it is true that placental CRH is identical to hypothalamic CRH in structure, immunoreactivity, and bioactivity, there is one major difference. In contrast to the inhibitory influence (negative feedback) on the promoter region of the CRH gene in the hypothalamus, maternal cortisol from the adrenal glands stimulates the placenta to express hCRHmRNA, establishing a positive feedback loop that allows for the simultaneous increase of CRH, ACTH, and cortisol over the course of gestation. The difference in behavior of the CRH gene in the placenta and hypothalamus is due to the expression of different transcription factors, co-activators, and co-repressors in these two tissues (King, Smith, & Nicholson, Reference King, Smith and Nicholson2002).
Concentrations of hCRHmRNA increase more than 20-fold in the weeks preceding delivery (Frim et al., Reference Frim, Emanuel, Robinson, Smas, Adler and Majzoub1990) resulting in exponentially rising elevations in maternal plasma CRH levels over the course of the second half of gestation (Campbell et al., Reference Campbell, Linton, Wolfe, Scraggs, Jones and Lowry1987; Chan et al., Reference Chan, Smith, Lewin, Brinsmead, Zhang, Cubis and Hurt1993; Goland et al., Reference Goland, Wardlaw, Stark, Brown and Frantz1986; Sasaki et al., Reference Sasaki, Shinkawa, Margioris, Liotta, Sato, Murakami and Yoshinaga1987; Wolfe et al., Reference Wolfe, Patel, Campbell, Anderson, Dornhorst, Lowry and Jones1988). The seminal study from Roger Smith's group (McLean et al., Reference McLean, Bisits, Davies, Woods, Lowry and Smith1995) determined that escalating levels of placental CRH (pCRH) are significantly linked to gestational length. They proposed that pCRH controlled a “placental clock” that regulates or alters the timing of parturition (McLean et al., Reference McLean, Bisits, Davies, Woods, Lowry and Smith1995; Smith, Mesiano, & McGrath, Reference Smith, Mesiano and McGrath2002). Elevated levels and steeper trajectories of pCRH initiate a cascade of events resulting in activation of smooth muscles in the uterine wall (myometrium) to induce contractions and expand the uterus to assist with delivery and in extreme cases contribute to preterm birth (Smith et al., Reference Smith, Smith, Shen, Engel, Bowman, McGrath and Smith2009; Wadhwa et al., Reference Wadhwa, Garite, Porto, Glynn, Chicz-DeMet, Dunkel-Schetter and Sandman2004). As illustrated in Figure 3, it is the trajectory of pCRH production (i.e., the rate of acceleration) over gestation, rather than the absolute hormone concentration, that best predicts preterm birth (delivery before 37 weeks gestation) suggesting that target cells are highly responsive to relative changes in pCRH concentrations (Smith et al., Reference Smith, Smith, Shen, Engel, Bowman, McGrath and Smith2009). Findings from our research illustrated in Figure 4, plotted from birth (a known time point) backward toward conception (an estimated time point) even more clearly shows the spike in CRH near delivery for the preterm birth contrasted with the women who delivered full term.

Figure 3. Concentration of pCRH over the course of gestation. The interesting finding from this study is that women delivering at term (N = 406) and preterm (N = 49) reach the same pCRH levels but at different rates.

Figure 4. Placental CRH concentrations plotted from the time of birth rather than gestation. The pattern of pCRH increase for women who deliver term is gradual as delivery approaches. For women who will deliver preterm, there is an abrupt spike in pCRH at delivery.
Other studies have demonstrated that the normal trajectory of placental CRH production over the course of gestation may be increased by an adverse intrauterine environment characterized by physiological stress. A series of in vitro studies by Petraglia and colleagues (Petraglia, Sawchenko, Rivier, & Vale, Reference Petraglia, Sawchenko, Rivier and Vale1987; Petraglia, Sutton, & Vale, Reference Petraglia, Sutton and Vale1989) have shown that CRH is released from cultured human placental cells in a dose-response manner to all the major biological effectors of stress, including cortisol, catecholamines, and pro-inflammatory cytokines. Moreover, CRH trajectories also are accelerated in high-risk conditions for preterm birth, such as pregnancy-induced hypertension, preeclampsia, fetal asphyxia, umbilical-placental vascular insufficiency, and multiple gestation (Giles, McLean, Davies, & Smith, Reference Giles, McLean, Davies and Smith1996; Goland et al., Reference Goland, Jozak, Warren, Conwell, Stark and Tropper1993; Goland, Conwell, & Jozak, Reference Goland, Conwell and Jozak1995). There is substantial in vitro and in vivo findings indicating that the placenta detects and responds to a variety of maternal stress signals and supports the assumption that levels of placental CRH are a direct index of fetal exposure to maternal stress. As discussed below, placental CRH may be an integrative pathway through which diverse prenatal stressors inform the fetus of the state of its environment and shape fetal developmental trajectories (Charil, Laplante, Vaillancourt, & King, Reference Charil, Laplante, Vaillancourt and King2010; O'Donnell, O'Connor, & Glover, Reference O'Donnell, O'Connor and Glover2009; Sandman, Davis, Buss, & Glynn, Reference Sandman, Davis, Buss and Glynn2011).
Fetal Programming: Is There a Role for Placental CRH?
The fetal programming (Barker, Reference Barker1998) and developmental origins of health and disease (Gluckman, Hanson, Cooper, & Thornburg, Reference Gluckman, Hanson, Cooper and Thornburg2008) models were based on retrospective observations that individuals born early or small for gestational age had higher than expected risk for poor health outcomes over their life span. Birth phenotype was believed to be a reflection of intrauterine “events” that placed the fetus at risk, but these earlier, seminal observations were primarily epidemiological and not designed to examine plausible causal pathways. An implicit assumption of contemporary modifications of the programming or developmental origins of health and disease models is that the fetus is an active participant in its own development (Sandman, Class, Glynn, & Davis, Reference Sandman, Class, Glynn, Davis and Rosenfeld2015; Sandman & Davis, Reference Sandman and Davis2012; Sandman, Davis, Buss, & Glynn, Reference Sandman, Davis, Buss and Glynn2012). It is argued that the human placenta, a fetal organ, collects information from the maternal environment and responds with a complex pattern of signals to the host. If the prenatal environment is perceived to be stressful or hostile (e.g., placental detection of increased levels of maternal cortisol), the fetal-placental signals (e.g., increased levels of CRH) to the mother may accelerate gestational trajectories resulting in preterm birth permitting fetal escape from an inhospitable environment and ensuring short-term survival (as described above and conceptually similar to the role of CRH in the accelerated metamorphosis of the tadpole). The fetus also incorporates this information to adjust its developmental program in preparation for survival after birth. Because the vulnerable fetal nervous system develops in a specific sequence from conception to birth, both the timing and the strength of maternal signals (e.g., cortisol in maternal response to stress) are critical factors that influence the neurodevelopmental outcomes for the fetus. If the fetus is the recipient of adverse, stressful, or chaotic patterns of maternal signals, the nervous system is remodeled in preparation for survival in a hectic postpartum environment (Sandman, Davis, & Glynn, Reference Sandman, Davis and Glynn2012). Remodeled tissue modifies the function and physiological capacity of the brain (and other organs as well) and is a fundamental assumption of how fetal exposures influence health and disease.
Placental CRH and fetal behavior
In a series of prospective studies to examine the influence of the intrauterine environment on the fetus, we have focused on the neurodevelopmental consequences of fetal exposure to patterns of psychobiological signals. We have examined the influence of both psychosocial and biological indicators of stress. Our research in over 900 maternal/offspring dyads has included outcomes in the human fetus, neonate, infant, toddler, and child including early adolescence. As discussed above, we have been particularly (but not exclusively) interested in pCRH because (a) it is a major stress peptide with direct effects on the nervous system and (b) deviations in pCRH provide objective evidence that the fetus has been exposed, and is responding to, variations of maternal signals of stress. Although there are many studies of CRH in animal models, our focus in pregnancy-related exposures and outcomes has been on humans because the expression of CRH in the placenta only has been reported in primates. In addition, because pCRH is reliably associated with gestational length (and in some studies birth weight; e.g., Stout, Espel, Sandman, Glynn, & Davis, Reference Stout, Espel, Sandman, Glynn and Davis2015), in all of our studies these two variables (as well as others) are included as covariates.
Assessment of the human fetal heart rate response (FHR) to startle and to habituation sequences provides a method for measuring neurological integrity before there are postpartum influences on development such as birth phenotype, socialization, and parenting (Sandman, Glynn, & Davis Reference Sandman, Glynn, Davis, Kisilevsk and Reissland2016; Sandman, Wadhwa, Hetrick, Porto, & Peeke, Reference Sandman, Wadhwa, Hetrick, Porto and Peeke1997). However, there are limitations to what can be achieved because of the narrow range of fetal behavior that can be measured, the obvious issue of access to the fetus, and the related issue of ethical constraints. Despite these limitations, we have examined the association between maternal concentrations of pCRH and fetal response to a startling, auditory challenge.
The structures of the human auditory system are well developed before birth. By 8 to 9 weeks gestation the cochlea is fully coiled and by 10 to 11 weeks gestation the hair cells begin to differentiate (Pujol & Lavigne-Rebillard, Reference Pujol and Lavigne-Rebillard1985). The development of the cochlea is completed by about 30 weeks gestation age (Pujol, Lavigne-Rebillard, & Uziel, Reference Pujol, Lavigne-Rebillard and Uziel1990) but the fetal auditory system (cochlea and brain stem) is capable of detecting and responding to sound by 25 weeks gestational age (Pujol et al., Reference Pujol, Lavigne-Rebillard and Uziel1990). Moreover, the neural circuitry of the startle response has been described in detail (Davis, Gendelman, Tischler, & Gendelman, Reference Davis, Gendelman, Tischler and Gendelman1982; Tischler & Davis, Reference Tischler and Davis1983). The startle response to an acoustic stimulus involves a pathway that synapses in the cochlear nucleus with further connections to the nucleus reticularis pontis caudalis (Wu, Suzuki, & Siegel, Reference Wu, Suzuki and Siegel1988). The activation at this nucleus is modulated by the amygdala, which confers emotional significance to the startling stimulus. Although the human fetus detects and responds to acoustic challenge by midgestation, it is unknown if the response is modulated by the amygdala.
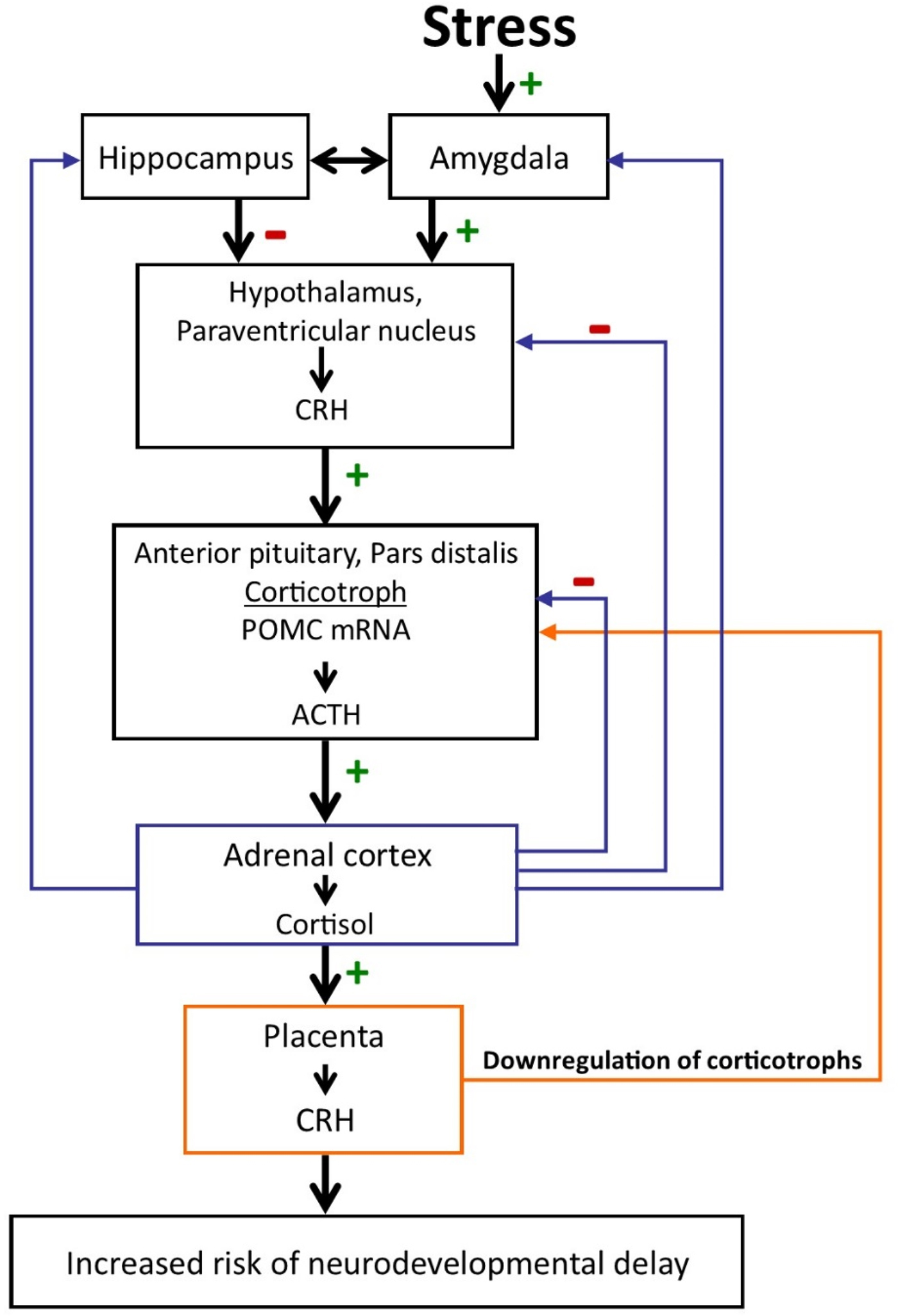
Figure 5. The regulation of the hypothalamic–pituitary–adrenal axis changes dramatically over the course of gestation with profound implications for the mother and the fetus. During pregnancy CRH is released from the placenta into both the maternal and the fetal compartments. In contrast to the negative feedback regulation of hypothalamic CRH, cortisol increases the production of CRH from the placenta. Placental CRH concentrations rise exponentially over the course of gestation. Elevated levels and/or rapid acceleration of CRH at various times during gestation is associated with increased risk of preterm birth, reduced growth, and neuromuscular development and neurodevelopmental delay. (+ signifies stimulation; - signifies inhibition.)
Fetal exposure to concentrations of pCRH is associated with fetal heart rate (FHR) responses to startle stimulation and to habituating stimulation. We determined that during vibroacoustic stimulation (VAS) the majority of fetuses respond to startle by 30 weeks gestation but only about half respond at 25 weeks (Buss et al., Reference Buss, Davis, Class, Gierczak, Patillo, Glynn and Sandman2009). Because there was evidence that some fetuses did respond at 25 weeks, we tested if these individual differences could be explained by exposures to placental markers of stress (Class et al., Reference Class, Buss, Davis, Gierczak, Pattillo, Chicz-DeMet and Sandman2008). We found that fetal exposure to the lowest concentrations of pCRH (i.e., bottom quartile) early in gestation (~15 weeks gestation) was associated with heightened fetal startle reflecting enhanced fetal maturity and accelerated neurological development. This partially confirmed the findings of an earlier and smaller study that assessed the effects of fetal exposure to pCRH on fetal memory and attention by presenting a series of repeated VAS, interrupted by a novel VAS. We found that lower placental CRH at 30 weeks gestation was associated with an improved ability of the fetus to habituate to repeated presentations of the VAS and to identify a novel stimulus (Sandman, Wadhwa, Chicz-DeMet, Porto, & Garite, Reference Sandman, Wadhwa, Chicz-DeMet, Porto and Garite1999).
A nearly identical influence of pCRH on fetal neurological maturation was observed in a habituation study that used pure tone simulation rather than the highly arousing VAS. FHR responses at 25, 30, and 36 weeks gestation were collected to 15 tones, followed by a novel tone (tone 16; dishabituation stimulus) and then 10 tones identical to the first 15 tones. As expected, the most efficient dishabituation was observed in the most mature fetuses, at 36 weeks gestation. However, the efficiency at 36 weeks was modulated by pCRH exposure earlier in gestation. Specifically, fetal exposure to the lowest level of pCRH at 15 weeks gestation was associated with the most efficient FHR dishabituation index (Sandman, Reference Sandman2015). Collectively, these findings suggested that exposure to CRH early in pregnancy influenced (programmed) fetal maturation as measured by startle and habituation: high concentrations “retard” maturation and low levels accelerate it.
It is tempting to conclude, or least speculate, that the association between exposure to pCRH at around 15 weeks gestation and the startle response later in gestation, at around 25 weeks, reflects an influence of pCRH on the acoustic pathway and perhaps on the amygdala that moderates this pathway. We have no evidence for the former possibility, but we have reported that human fetal exposure to elevated cortisol (which stimulates the synthesis and released of pCRH) at 15 weeks gestation is associated with increased amygdala volume in 7-year-old children (Buss et al., Reference Buss, Davis, Shahbaba, Pruessner, Head and Sandman2012). Moreover, repeated or severe stress in an immature rodent results in increased expression of CRH mRNA in the amygdala (Avishai-Einer, Brunson, Sandman, & Baram, Reference Avishai-Eliner, Brunson, Sandman and Baram2002; Hatalski, Gourguis, & Baram, Reference Hatalski, Guorguis and Baram1998). Furthermore, in the immature rat, CRH responses to stressful events are abundantly present even though there may be limited response from the pituitary or adrenal gland (Vazquez et al., Reference Vazquez, Bailey, Dent, Okimoto, Steffek, Lopez and Levine2006). It is fair to conclude that it is not known if exposure to pCRH influences the human fetal startle response at a critical period in the development of the acoustic pathway, but there is inferential support for an influence on the amygdala. For instance, there is both expression and activity of CRH in response to stress in the immature rodent nervous system coupled with the indirect evidence in children that a primary stimulus for increased pCRH synthesis and release during early gestation is associated with increased volume of the amygdala. Therefore, it is plausible that human fetal exposure to pCRH at 15 weeks gestation can influence the volume of the amygdala and therefore its function as a modulator of the acoustic system. However, as we will see below, the association between the timing of fetal exposure to pCRH and the structure of the nervous system may be much more complex.
Placental CRH and physical growth and development
In addition to the key role pCRH plays in the regulation of gestational length and aspects of fetal maturation, it may contribute to early parameters of growth and physical development. In a group of 246 women and their healthy children we (Stout et al., Reference Stout, Espel, Sandman, Glynn and Davis2015) found that elevated pCRH at 30 weeks gestation was associated both with reduced body mass index and weight at birth (adjusted for gestational age). Significant patterns of catch-up growth from 3 to 24 months was associated with elevated pCRH levels. Small size at birth coupled with catch-up growth is associated with increased risk for subsequent obesity. There are several plausible pathways by which pCRH may affect fetal growth, body composition, and physical maturity. One possibility is that increasing levels of pCRH leads to fetal nutrient restriction, resulting in a “thrifty” phenotype (Gangestad, Caldwell Hooper, & Eaton, Reference Gangestad, Caldwell Hooper and Eaton2012). Nutrient-related restricted fetal growth may result in preparation (programming) for a nutrient-deprived postnatal environment. A mismatched nutrient-rich postpartum environment can induce a “thrifty” phenotype resulting in obesity and metabolic risk (Hales & Barker, Reference Hales and Barker2001).
In addition to size, neuromuscular and physical characteristics (the New Ballard Maturation Protocol) were evaluated in 158 newborn children within 24 hr of birth (M = 8.9 hr) before the possibility of postpartum influences (Ellman et al., Reference Ellman, Dunkel-Schetter, Hobel, Chicz-DeMet, Glynn and Sandman2008). Newborns exposed to elevated pCRH at 30 weeks gestation exhibited significant decreases in measures of muscle tone, distinct posture, angles of resistance in key muscle groups, and in maturation of skin, lanugo, plantar surface of the foot, breast, eye/ear, and genitals. Although neuromuscular maturation and body size are key factors influencing adaptation, it is not entirely clear how reduced growth and delayed neuromuscular maturation are causally connected except that they are both associated with elevated prenatal levels of pCRH at 30 weeks.
Placental CRH, gestational timing, and brain development
As discussed above, pCRH levels continue to increase throughout gestation until delivery and then fall within 24 hr. It is reasonable to assume that the timing of fetal exposure to pCRH coupled with the timetable for brain development collaborate to determine the programmed effect. Our studies suggest that fetal neural maturation is influenced primarily by pCRH levels early in gestation, around 15 weeks. Gestational length is associated with accelerated trajectories of pCRH between 20 and 25 weeks and growth and physical maturation with pCRH levels at 30 weeks. We have invested in studies of gestational length, fetal behavior, and factors related to growth to lay the groundwork for our primary interest: the long-term effects of fetal exposures to psychobiological stress on the brain and behavior.
The human brain is the most complicated organ in living systems and is undergoing massive development during fetal life. This structure originates from a simple neural tube, followed by a series of differentiation processes over a long period of time including the entire fetal period and into early adulthood. There are critical periods of neural migration in early human development when the nervous system is especially vulnerable to disruptions. Radial neuronal cell migration begins in the human brain around 42 days gestational age (Stiles & Jernigan, Reference Stiles and Jernigan2010; Takahasi, Folkerth, Galaburda, & Grant, Reference Takahashi, Folkerth, Galaburda and Grant2012). Between gestational age 8 and 16 weeks, migrating neurons forming the subplate zone receive connections from afferent neurons originating in the thalamus, basal forebrain, and brainstem. Concurrently, cells accumulating in the outer cerebral wall form the cortical plate, which eventually will become the cerebral cortex. By gestational week 20, there is an exponential increase in cortical thickness (Huang et al., Reference Huang, Xue, Zhang, Ren, Richards, Yarowsky and Mori2009), axons form synapses with the cortical plate, and by gestational week 24, cortical circuits are organized (Kostovic, Judas, Rados, & Hrabac, Reference Kostovic, Judas, Rados and Hrabac2002; Bourgeois, Goldmanrakic, & Rakic, Reference Bourgeois, Goldmanrakic and Rakic1994). By gestational week 28, the fetal brain is forming secondary and tertiary gyri, and exhibiting neuronal differentiation, dendritic arborization, axonal elongation, synapse formation and collateralization, and myelination1 (Bourgeois et al., Reference Bourgeois, Goldmanrakic and Rakic1994).
In the first test of the long-term behavioral consequences of human fetal exposure to pCRH, we found that elevations in midgestation (at 25 weeks) pCRH was linked to increased maternal report of fear and distress in 2-month-old infants (N = 246; Davis et al., Reference Davis, Glynn, Dunkel-Schetter, Hobel, Chicz-DeMet and Sandman2005). Levels of pCRH at earlier (19 weeks) and later (31 weeks) intervals were not related to infant temperament. Although there were adjustments made for maternal bias, the findings still relied on mother's report. Recently we tested the same general question in a group of 91, 5-year-old children with self-reported measures collected in a structured interview (Howland, Sandman, Glynn, Crippen, & Davis, Reference Howland, Sandman, Glynn, Crippen and Davis2016). All children and their mothers had been evaluated during gestation with the complete battery of measures including pCRH. We found that elevated pCRH around 23 weeks (using hierarchical linear model estimates) significantly was associated with increased reports of internalizing symptoms. These findings suggest that fetal exposure to pCRH is associated with a specific long-term profile of behavior that may be linked to neural events occurring around 25 weeks of gestation.
Placental CRH, behavior, and brain volume in children
Enduring alterations of brain structure and function at molecular, cellular, and circuit levels (Peña et al., Reference Peña, Kronman, Walker, Cates, Bagot, Purushothaman and Nestler2017) are considered fundamental mechanisms of how early life experiences influence health and disease (Bale et al., Reference Bale, Baram, Brown, Goldstein, Insel, McCarthy and Nestler2010; McMullen et al., Reference McMullen, Langley-Evans, Gambling, Lang, Swali and McArdle2012). To assess the possibility that exposure to pCRH influences brain structure, we determined the association between prenatal exposure to pCRH throughout gestation and brain volume in 97 typically developing 6- to 9-year-old children (Sandman et al., Reference Sandman, Curran, Davis, Glynn, Head and Baram2018). Levels of pCRH had been determined from maternal blood during fetal development at 15, 19, 25, 31, and 36 weeks gestation. Brain volume/thickness was measured with structural magnetic resonance imaging. Assessment of children's internalizing and externalizing behaviors were determined from structured interviews administered to mothers. Inhibition and attention were assessed in the children with neuropsychological tests. We found that elevated levels of pCRH averaged across gestation were associated with significant thinning in 12% of the cortex distributed equally in the left and right hemispheres. As presented in Figure 6, thinning associated with pCRH levels was most prominent in the temporal and frontal regions.
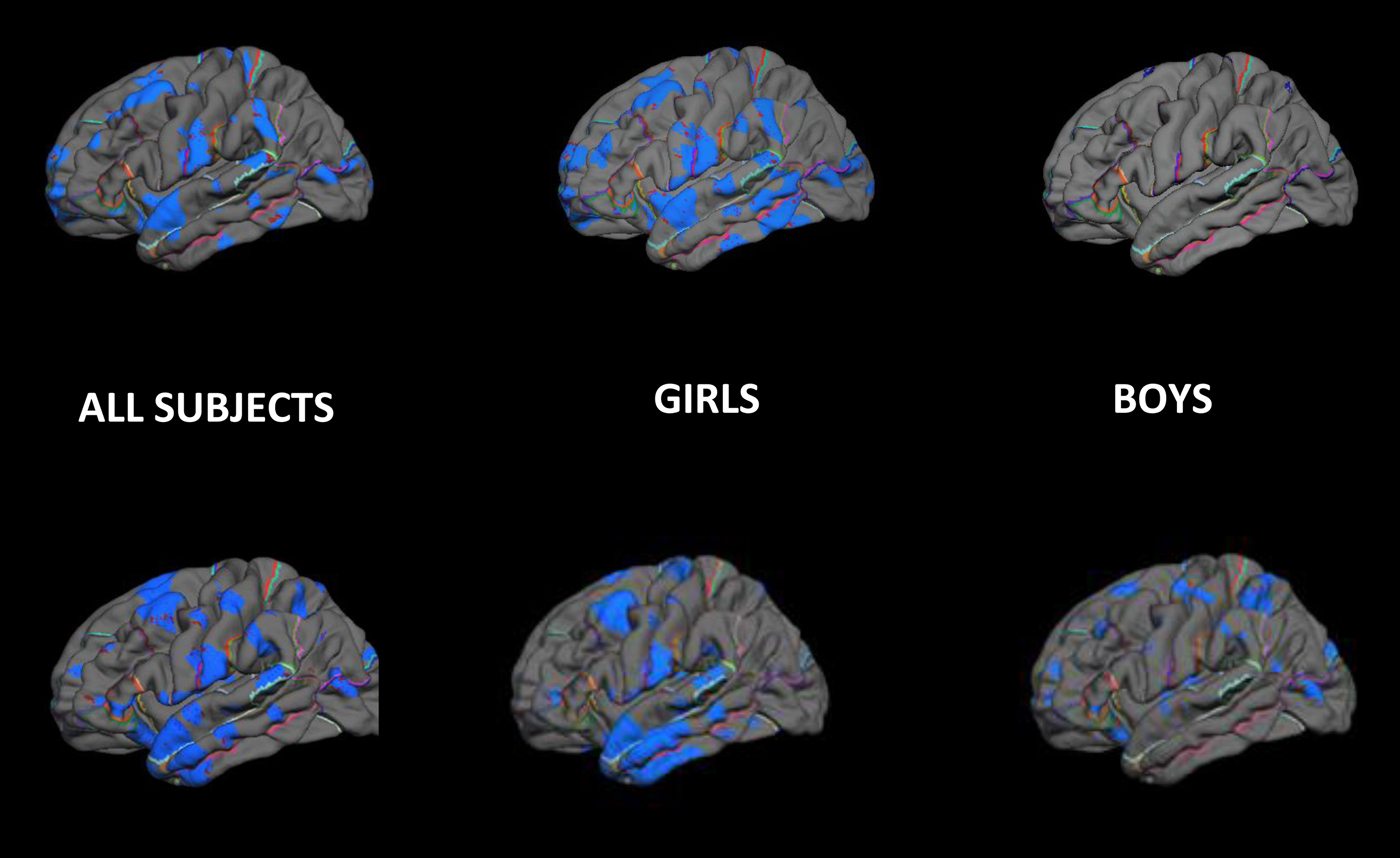
Figure 6. Pial maps illustrating cortical areas in children that have significant (negative) associations with fetal exposure to pCRH. Areas in blue reflect that thinner cortical mass is associated with higher levels of fetal exposure to pCRH. The effects observed in the total sample from both fetal exposures at 19 weeks and 31 weeks primarily are the result of the strongest association in girls.
We then examined the influence of gestational timing of pCRH exposure on cortical thickness. We found that fetal exposure to pCRH early in gestation (15 and 19 weeks) was associated with localized thinning bilaterally in the frontal poles (69% of the structure in the left and 78% in the right). A mediational model supported the argument that reduced cortical volume in the frontal pole, associated with elevated fetal exposure to concentrations of pCRH at 19 weeks’ gestation, contributed to externalizing symptoms in 6- to 9-year-old children. Exposure to pCRH later in gestation, at 25 and 31 weeks, was associated with cortical thinning bilaterally in the temporal poles. Remarkably, 98% of the lateral surface of the right temporal pole and 66% of the left temporal pole were significantly thinner in children exposed to high levels of pCRH at 31 weeks gestation. A mediational model indicated that reduced right temporal pole volume associated with pCRH contributed to poorer performance on a visual processing and a sustained attention task.
The effects of pCRH on brain volume at both 19 and 31 weeks were observed primarily in girls. The association between fetal exposure to pCRH at 19 weeks’ gestation and cortical thinning involved most cortical areas in girls but minimal areas in boys (see Figure 6). Fetal exposure to pCRH at 31 weeks affected cortical thinning globally in boys but locally in the temporal pole in girls; similar to the findings for the combined sexes (Figure 6). It is critical to note that there was no evidence that prenatal exposure to pCRH at any gestational interval was associated with increased cortical thickness.
These findings of an association between fetal exposure to pCRH and brain volume are suggestive, but direct effects of the peptide on brain development cannot be concluded. For instance, a third variable or general stress-related mechanism could be responsible for the observed findings. In a companion study (Curran, Sandman, Davis, Glynn, & Baram, Reference Curran, Sandman, Davis, Glynn and Baram2017), we used a rodent model to test if CRH has direct effects on brain structure. Cortical neurons were grown in cultures with graded concentrations of CRH. Exposure to CRH resulted in a significant, dose-dependent impoverishment of the branching of pyramidal-like cortical neurons. These results are consistent with the possibility that CRH directly decreases dendritic branching. Because cortical volume is largely made up of dendritic trees of cortical neurons, reduction in dendritic arborization may be measured as volume loss in magnetic resonance imaging. Thus, it is plausible that fetal exposure to high levels of pCRH has direct effects on cortical volume by reducing dendritic complexity and thereby contributes to the behavioral consequences observed as children develop.
Conclusion
The questions posed at the beginning of this paper have been answered. “What is CRH?” CRH is a 41 amino acid peptide hormone that resides in a 172-residue bioinactive precursor molecule. The gene coding for CRH is located on the long arm of chromosome 8 and is expressed throughout the nervous system and in peripheral sites. The search for “Where does CRH come from?” generated a new way to think about the brain: the brain as an endocrine organ. CRH was not the first releasing hormone discovered in the brain, but the belief that it must exist to regulate the stress response was the catalyst for changing endocrinology forever. CRH is liberated from its precursor by enzymes localized in the paraventricular nucleus of the hypothalamus. It travels down a portal system to the median eminence where it is stored before acting on the anterior pituitary gland to stimulate the syntheses of POMC. The focus of this paper is pCRH. Its discovery in the placenta is as astonishing as is the discovery of CRH in the brain; however, its placental origin has not been accorded the same acknowledgment. As with the brain, the placenta has become recognized as an endocrine organ. Placental CRH is identical to hypothalamic CRH in every way except for the positive feedback with cortisol. Cortisol stimulates the synthesis and release of CRH in the placenta unlike the negative feedback it has in the hypothalamus.
The last question: “Does CRH have anything to do with human development?” has been answered. Placental CRH is synthesized and released in response to many, if not all, biological and psychosocial stressors and to health conditions that threaten fetal survival and is among the most salient signals shaping the human fetus. Just as release of hypothalamic CRH increases the probability that the tadpole will survive an inhospitable environment by accelerating development, pCRH release in humans advances the placental clock for an early delivery. The surviving toad exposed to elevated CRH is small and immature. The same is true for preterm children with well-documented developmental consequences, but even more important are the small and physically immature children born at term but exposed prenatally to elevated pCRH. It is reasonable to conclude that fetal exposure to pCRH, independent from birth outcomes, has significant consequences for human growth and neurodevelopment. Moreover, the human fetus exposed to elevated pCRH has arousal and learning profiles that may be prodromal to later abilities that make adaption to environmental challenges more problematic. The findings of negative temperament and neuropsychiatric symptoms in children exposed to pCRH may make them less desirable romantic partners with fewer reproductive opportunities as adults.
In concert with the companion finding in the rodent, the magnetic resonance imaging results with children suggest that the immature nervous system is remodeled by exposure to pCRH. From these findings, we can argue that the associations observed between exposure to pCRH and behavior may be causally mediated by changes in the brain. We reported that a plausible mechanism for the decreased cortical volume in children exposed to prenatal CRH was diminished density and complexity of dendritic trees. These findings of enduring neural consequences for fetal exposure to pCRH provide one explanation for how early life adversity can influence health and well-being. Moreover, the different cortical volume profiles associated with timing of pCRH exposure provide support for the behavioral differences associated with gestational exposure. Clearly the exposure–structure–function associations need to be further investigated.
There is evidence that there are different consequences for males and females exposed to pCRH. We reported this as a viability–vulnerability trade-off (Sandman, Glynn, & Davis, Reference Sandman, Glynn and Davis2013). Briefly, sex differences in response to stress have been observed in mammalian animal models as early as meiosis (Hunt & Hassold, Reference Hunt and Hassold2002). For instance, when faced with adversity (a toxin), male meiosis is interrupted resulting in infertility. However, a similar adversity does not interrupt meiosis in females resulting in greater chances of survival but with the possible risk of subsequent chromosomal abnormalities. Moreover, within weeks of implantation, the female placenta is more responsive than the male placenta to changes in stress signals including detection and response to maternal glucocorticoid concentration (Clifton, Reference Clifton2010). It also has been argued that because male fetuses invest resources in growth, they do not have adequate reserves to adapt to maternal signals (such as glucocorticoid concentrations) and have limited ability to adjust to adversity, placing them at greater risk for subsequent mortality and neurological morbidity. Because of their failure to make adjustments to environmental signals, males are more vulnerable to severe consequences of early life stress such as preterm birth (Cooperstock & Campbell, Reference Cooperstock and Campbell1996), have poorer neonatal and infant health outcomes (Peacock, Marston, Marlow, Calvery, & Greenough, Reference Peacock, Marston, Marlow, Calvert and Greenough2012), are less likely to survive in intensive care, and are at higher risk for developmental delay (Aiken & Ozanne, Reference Aiken and Ozanne2013). We argued that the surviving males consist of a culled “homogenous” cohort in contrast with the more diverse females who escape severe early consequences because of their adaptive agility. Because of this inherent variability in females, the probability is increased that there will be significant associations between early life events and their developmental trajectories.
Except for the risk of preterm birth, the consequences of elevated pCRH for the mother was not discussed. There are other consequences for the mother, including risk for postpartum depression (Glynn & Sandman, Reference Glynn and Sandman2014; Yim et al., Reference Yim, Glynn, Dunkel-Schetter, Hobel, Chica-DeMet and Sandman2009). As reported above, in contrast to the negative feedback regulation of hypothalamic CRH, cortisol stimulates the expression of pCRH, which establishes a positive feedback loop resulting in the simultaneous increase in pCRH, ACTH, b-endorphin, and cortisol over the course of gestation. Increased circulating pCRH may downregulate the maternal corticotrophs, reducing responses to internal signals, suppressing the secretion of hypothalamic CRH in the second half of pregnancy and in the immediate postpartum. If suppression of CRH persists, it may create a central CRH deficiency and increase the risk for depressive symptoms (Glynn & Sandman, Reference Glynn and Sandman2014).
The importance of considering gestational timing cannot be underestimated, for both exposure to maternal stress and the development of brain systems. Signals from the maternal stress systems are continuous sources of information to the fetus over the course of gestation. Levels of pCRH are detected by the eighth week of pregnancy and increase geometrically over the last trimester. Depending on the severity and the timing of maternal signals and the timetable of organogenesis, the sequence of neural development can be disrupted, resulting in programmed consequences for brain structure and behavior. We have found that there are unique associations between exposure to pCRH at different gestational intervals and measures of later brain structure and behavioral outcomes. Studies of prenatal influences on development that only include a single time point may miss a critical or sensitive period for observing important associations with specific outcomes. For instance, exposure to pCRH early in gestation was associated with a different pattern of brain thinning than later exposures. In this case, examination of a single time point would result in an incomplete and inaccurate understanding of the neurological significance of prenatal exposure to pCRH. Moreover, we reported here, and confirmed elsewhere, that analysis of patterns of maternal signals, requiring multiple time points, during gestation are sometimes far superior predictors of birth and child outcomes than level or severity at a single time point (Davis & Sandman, Reference Davis and Sandman2010; Glynn, Dunkel-Schetter, Hobel, & Sandman, Reference Glynn, Dunkel-Schetter, Hobel and Sandman2008; Kane, Dunkel-Schetter, Glynn, Hobel, & Sandman, Reference Kane, Dunkel-Schetter, Glynn, Hobel and Sandman2014; Sandman, Davis, Buss, et al., Reference Sandman, Davis, Buss and Glynn2012).
There is one major caveat that is important to consider before accepting the conclusions about the independent actions of CRH/pCRH on gestation, fetal development, and on the brain and behavior. The complex interactions among CRH, pCRH, and other molecules, especially cortisol, and psychosocial and demographic variables, were not described in sufficient detail. We understand the importance of these associations and interactions and most of the studies from our program included their consideration, but it would be beyond the focused scope of this paper to include all that material. The findings presented here for CRH and pCRH survived after controlling or considering these relevant factors.






