In 2011, when Dr. Cicchetti invited us to write this paper, he encouraged us
… to set out the future agenda for research, translation, and policy in the decades ahead. . . . I want your paper to be forward-thinking and visionary in ambition. I am not asking you to provide a systematic review article; rather, I am seeking a contribution that is conceptual, opinion driven, and innovative: one that provides a blueprint for future empirical research, translational research, and policy implications. (D. Cicchetti, personal communication, June 15, 2011)
We have written this paper for behavioral researchers and clinicians interested in development rather than for experts in genomics. Following the Editor in Chief's advice, we have tried to make the paper conceptual and opinion driven, rather than provide a systematic review. Our goal was to discuss a few issues that will shape the future of developmental research and, eventually, translate to the clinic. No crystal ball is needed to see some of the future of genomics because the breathtaking momentum of current advances will sweep the field into the next decade. However, genomics is moving so fast that we can confidently predict that some important advances will be completely new and unanticipated.
We waited until 2013 for three reasons. First, we wanted to contribute to this 25th anniversary issue of the Journal, which has promoted genetic research in development over the decades by publishing more than 100 papers on genetics, even early on when genetics was not as popular as it is now. Second, we wanted to wait until the publication in November 2012 of this Journal's Special Issue, “Genomic Sciences for Developmentalists: The Current State of Affairs” (Grigorenko & Cicchetti, Reference Grigorenko and Cicchetti2012). This Special Issue provides reviews of research in genetics and genomics and shows the great extent to which developmental psychopathologists have increasingly incorporated genomics in their research. The Special Issue frees us to focus on the future because it covers background issues to which we refer along the way; more details about these background issues can be found elsewhere (Plomin, Reference Plomin2013; Plomin, DeFries, Knopik, & Neiderhiser, Reference Plomin, DeFries, Knopik and Neiderhiser2013). We also provide a glossary at the end of this paper so that we can avoid definitions in the text; we denote terms included in the glossary with boldface type, beginning with genetics and genomics. Third, we wanted to wait until the next big thing, whole-genome sequencing, began to materialize through the dust settling from the recent explosion of research on genomewide association (GWA) studies using DNA arrays.
What Is Inherited Is DNA Sequence Variation: Everything Else Is a Phenotype
We begin with a general conceptual issue about DNA that is critical for understanding the future role of genomics in developmental research and translation. In this paper, we focus on DNA (genetics and genomics) because that is what is inherited. Specifically, we focus on less than 1% of the 3 billion base pairs of DNA that differ from person to person. This DNA sequence variation is responsible for all inherited physical, physiological, and psychological differences between individuals. Although the 99% of DNA that does not differ between individuals is vitally important for development, we focus on what makes individuals differ. All of the other “-omics” in between genomics and behavior are important endophenotypes for understanding pathways between genes and individual differences in outcomes, but they are not inherited by offspring (see Figure 1). For example, many mechanisms affect the extent to which DNA is transcribed into RNA in response to internal and external environments, such as adding methyl groups to DNA (epigenomics) or other mechanisms of gene expression (transcriptomics). Several papers in the special genomics issue of this Journal reported research on epigenetics and epigenomics (Bick et al., Reference Bick, Naumova, Hunter, Barbot, Lee and Luthar2012; Knopik, Maccani, Francazio, & McGeary, Reference Knopik, Maccani, Francazio and McGeary2012; Monk, Spicer, & Champagne, Reference Monk, Spicer and Champagne2012) and transcriptomics (Carlyle et al., Reference Carlyle, Duque, Kitchen, Bordner, Coman and Doolittle2012; Naumova et al., Reference Naumova, Palejev, Vlasova, Lee, Rychkov and Babich2012). After DNA is transcribed into RNA, some RNA is translated into amino acid sequences that produce our entire complement of proteins (proteomics). One of the most exciting findings in recent years is about the “dark matter” of DNA, which refers to the more than 98% of the genome that is not translated into amino acid sequences and had been thought to be “junk.” We now know that more than half of this DNA can be expressed in the sense that it is transcribed into RNA although it is not translated into amino acid sequences; the RNA itself can be functional in the sense of regulating the expression of other genes (Pennisi, Reference Pennisi2012).

Figure 1. (Color online) All of the “-omic” pathways between the genome and behavior are important for understanding individual differences. However, DNA sequence variation in the genome is ultimately responsible for hereditary influences on behavior. Correlations between DNA sequence variation and behavior are causal, as indicated by the single-headed arrow from the genome to behavior. Although behavior is often referred to as the observable “phenome,” all levels between the genome and behavior can be considered as phenotypes, which we show as connected by double-headed arrows signifying possible two-way directions of effect. The genetic and environmental origins of individual differences in these phenotypes need to be assessed rather than assumed.
Although the ultimate goal is to understand all the mechanisms by which DNA sequence variation contributes to individual differences in behavioral development, all of these processes downstream from DNA can be viewed as phenotypes. For example, individual differences in the amount of methylation across the genome (epigenomics) or individual differences in the number of RNA transcripts for all transcribed DNA in the genome (transcriptomics) are phenotypes that are age specific, state specific, and tissue specific. The genetic and environmental origins of individual differences in these processes need to be assessed rather than assumed, like any other phenotype. Often discussed as a possible exception to this rule (i.e., all that is inherited is DNA sequence variation) is the inheritance of epigenetic markings from parent to offspring (Slatkin, Reference Slatkin2009). For example, some methylation markings that silence genes in mothers may be transmitted to their offspring (Hackett et al., Reference Hackett, Sengupta, Zylicz, Murakami, Lee and Down2013). However, the extent of transmitted methylation seems limited because as sperm and eggs develop, markings from the parent are erased, and a further clean-up job happens when the egg is fertilized. Only a few markings fail to be erased, and those that slip through might not matter. Empirically, twin studies suggest that individual differences in DNA methylation appear to be largely environmental in origin rather than inherited (Kaminsky et al., Reference Kaminsky, Tang, Wang, Ptak, Oh and Wong2009; Wong et al., Reference Wong, Caspi, Williams, Craig, Houts and Ambler2010; Wong et al., Reference Wong, Meaburn, Ronald, Price, Jeffries and Schalkwyk2013). Epigenetics may be an especially good biomarker of early developmental adversity (Bell & Spector, Reference Bell and Spector2011; Gordon et al., Reference Gordon, Joo, Powel, Ollikainen, Novakovic and Li2012; Heijmans & Mill, Reference Heijmans and Mill2012; Knopik et al., Reference Knopik, Maccani, Francazio and McGeary2012; Monk et al., Reference Monk, Spicer and Champagne2012). Again, we emphasize that all of these processes between genes and behavior are important in their own right. However, all that is inherited is DNA sequence variation: everything else is a phenotype.
For this reason, DNA sequence variation is in a causal class of its own in the sense that there is no direction of effects issue when it comes to correlations between genes and behavior. That is, correlations between DNA sequence variation and behavior are ultimately causal from genes to behavior because our behavior and experiences do not change DNA sequence variation. Other correlations between behavior and biology, including all the -omics and the brain, are questionable concerning the direction of effects, that is, whether the correlation is caused by the effects of behavior on biology or vice versa. As a result, one of the great strengths of DNA sequence variation is that it can be used to predict problems long before they appear, even prenatally. This predictive ability will enable research on interventions to prevent disorders rather than trying to reverse a disorder after it appears and causes collateral damage. The ultimate hope is for personalized genomics, individualized gene-based diagnoses, and treatment programs (Collins, Reference Collins2010).
The Future Includes Quantitative Genetics
Quantitative genetics might seem to belong to the past rather than the future. Quantitative genetics refers to designs such as family, twin, and adoption studies in humans and inbred strains and selection studies in nonhuman animals that estimate the overall extent to which the genome and the environome account for individual differences in a trait without knowledge of specific genetic or environmental factors. These designs are called quantitative genetics, not because they are limited to quantitative dimensions (these methods can also be applied to qualitative disorders) but because they assume that many genes are responsible for genetic influence (polygenic). Quantitative genetics is often contrasted with molecular genetics, a term that has been used broadly to refer to research using DNA. Until the last two decades, these two worlds had been growing apart since their births a century ago. In relation to inheritance, the goal of molecular genetics was to identify and understand genes, and the field focused on single-gene mutations of very large effect. The genes responsible for most of the thousands of monogenic disorders have been identified, and molecular geneticists have turned to complex traits and common disorders that are highly polygenic. Meanwhile, quantitative geneticists had not attempted to identify the genes responsible for genetic influence on complex traits because single-gene research strategies seemed inappropriate for what was assumed to be highly polygenic traits. A momentous shift is underway; these two worlds of genetics are merging as genomic technology makes it possible to identify the polygenic origins of individual differences in complex traits and common disorders.
Nearly all that we know with certainty about the genetics of the development of complex disorders and dimensions comes from quantitative genetic research designs, such as family, twin, and adoption studies. We predict that quantitative genetic studies will become increasingly important in the era of molecular genetics as these two worlds of genetics merge (Haworth & Plomin, Reference Haworth and Plomin2010), a prediction supported by the fact that the number of twin studies around the world has doubled during the past decade (Hur & Craig, Reference Hur and Craig2013). Quantitative genetic research has gone well beyond the rudimentary discovery of the ubiquitous and powerful influence of genes. Although it is an important first step to ask whether and how much genes affect behavioral dimensions and disorders (heritability), the key developmental question is how: how genotypes become phenotypes. For example, longitudinal studies have revealed genetic change as well as continuity in development, multivariate studies have found a surprising degree of genetic overlap as well as specificity between traits, and studies of gene–environment interplay have demonstrated the importance of gene–environment correlation as well as interaction (Plomin, DeFries, et al., Reference Plomin, DeFries, Knopik and Neiderhiser2013). One major direction for growth is the application of quantitative genetic research designs, primarily twin studies, to individual differences at all the -omic levels between genes and behavior (van Dongen, Slagboom, Draisma, Martin, & Boomsma, Reference van Dongen, Slagboom, Draisma, Martin and Boomsma2012). The twin design is uniquely powerful for understanding not only the genetic and environmental etiologies of the variance at each level but, importantly, the covariance between levels. The effort in launching such large twin studies insures that most of them continue longitudinally, making them especially valuable for developmentalists. Although the “and environmental” phrase has often been ignored except as the residual variance not explained by genetics, more and more research is taking advantage of the power of the twin method to investigate the environment. For instance, developmental studies increasingly use genetically sensitive designs such as the twin method to identify causal environmental effects free of genetic mediation (Harold, Elam, Lewis, Rice, & Thapar, Reference Harold, Elam, Lewis, Rice and Thapar2012; Jaffee & Price, Reference Jaffee and Price2012), and epigenomic studies use discordant monozygotic (MZ) twins to identify biomarkers of nonshared environmental influences that make genetically identical co-twins different, as indicated earlier.
If we could identify all of the genes responsible for heritability, there would be no more need for quantitative genetic research. However, quantitative genetic research will continue to make important contributions because we are a long way from identifying all of the genes responsible for the heritability of any complex trait. This is the “missing heritability” problem of GWA studies, discussed later, in which heritability is defined by quantitative genetics. Quantitative genetics will continue to be an important part of the future of developmental research for three reasons. The most general reason is that quantitative genetics is genomic. That is, quantitative genetic designs appraise the net effect on traits of genes throughout the genome regardless of the types of genes (e.g., structural variants or noncoding genes in addition to traditional coding genes), the number of genes, the frequency of their alleles, the size of their effects, or the complications of their interactions with other genes or environments; these are the culprits thought to be responsible for missing heritability. The second reason is that quantitative genetics is as much about the environment as it is about genetics. Because heritability is always substantially less than 100%, quantitative genetic research provides the best available evidence for the importance of the environment, while controlling for the environment. Because heritability is substantial, environmental studies can exploit genetically sensitive designs to investigate the environment, while controlling for possible confounding genetic influence, and to explore the developmental interplay between the environment and genes.
Quantitative Genetics Using DNA Alone
The third reason why quantitative genetics will continue to be important to developmentalists is a new technique that estimates the net effect of genetic influence using DNA alone in unrelated individuals rather than using familial resemblance in groups of special family members, such as MZ and dizygotic (DZ) twins who differ in genetic relatedness (Zaitlen & Kraft, Reference Zaitlen and Kraft2012). The novelty and importance of this technique for future quantitative and molecular genetic research leads us to describe this advance in some detail.
Like other quantitative genetic techniques, these new DNA-based methods use genetic similarity to predict phenotypic similarity. However, instead of using genetic similarity from groups differing markedly in genetic similarity, such as MZ and DZ twins, DNA-based methods use genetic similarity for each pair of unrelated individuals based on that pair's overall similarity across hundreds of thousands of single nucleotide polymorphisms (SNPs); each pair's genetic similarity is then used to predict their phenotypic similarity. Even remotely related pairs of individuals are excluded (up to fifth-degree relatives whose genetic similarity is 3%) so that chance genetic similarity is used as a random effect in a linear mixed model. Unlike MZ and DZ twins, who are 100% and 50% similar genetically, the chance DNA similarity between pairs of unrelated individuals only varies from –2% to +2% (see Figure 2). Nonetheless, overall genetic influence can be estimated by the extent to which this random DNA similarity can predict phenotypic similarity.
Figure 2. (Color online) The starting point for Genome-Wide Complex Trait Analysis (GCTA) is a matrix of chance genetic similarity across hundreds of thousands of single nucleotide polymorphisms pair by pair for large samples of unrelated individuals. Adapted from “Common DNA Markers Can Account for More Than Half of the Genetic Influence on Cognitive Abilities,” by R. Plomin, C. M. A. Haworth, E. L. Meaburn, T. Price, Wellcome Trust Case Control Consortium 2, and O. S. P. Davis, Reference Plomin, Haworth, Meaburn, Price and Davis2013. Psychological Science. Advance online publication. Copyright 2013 by the Association for Psychological Science. Adapted with permission.
In contrast to the twin method, which only requires a few hundred pairs of twins to estimate moderate heritability and does not need DNA, the demands of the DNA-based method are daunting: it requires not only DNA but also genotyping hundreds of thousands of SNPs for many thousands of individuals. Samples of many thousands are required because the method attempts to extract a slight signal of genetic similarity from the massive noise of hundreds of thousands of SNPs. The power of the method comes from comparing not just two groups, like MZ and DZ twins, but millions of pairs of individuals. For example, a sample of 3000 individuals provides nearly 5 million pair-by-pair comparisons. The first application of this approach was included in a software package with the clever acronym GCTA, which stands for genomewide complex trait analysis (Yang et al., Reference Yang, Benyamin, McEvoy, Gordon, Henders and Nyholt2010; Yang, Lee, Goddard, & Visscher, Reference Yang, Lee, Goddard and Visscher2011). We will refer to this DNA-based quantitative genetic method as GCTA, although other names have been proposed, such as linear mixed model and genomic-relatedness matrix restricted maximum likelihood; other methods (So, Li, & Sham, Reference So, Li and Sham2011) and modifications (Speed, Hemani, Johnson, & Balding, Reference Speed, Hemani, Johnson and Balding2012) are also emerging (Zaitlen & Kraft, Reference Zaitlen and Kraft2012; Zhou, Carbonetto, & Stephens, Reference Zhou, Carbonetto and Stephens2013).
Despite the daunting demands of GCTA for hundreds of thousands of SNPs genotyped on many thousands of individuals, hundreds of data sets already exist that meet these requirements because these are the same requirements for GWA studies, and these data sets are now being used to conduct GCTA. GCTA research across the life sciences generally yields evidence for significant genetic influence, confirming the results of twin studies. These include GCTA reports on common medical diseases (Lee et al., Reference Lee, Harold, Nyholt, Goddard, Zondervan and Williams2013), psychopathology (Lee, DeCandia, et al., Reference Lee, DeCandia, Ripke and Yang2012; Lee, Wray, Goddard, & Visscher, Reference Lee, Wray, Goddard and Visscher2011; Lubke et al., Reference Lubke, Hottenga, Walters, Laurin, de Geus and Willemsen2012), cognitive abilities (Benyamin et al., Reference Benyamin, Pourcain, Davis, Davies, Hansell and Brion2013; Davies et al., Reference Davies, Tenesa, Payton, Yang, Harris and Liewald2011; Deary et al., Reference Deary, Yang, Davies, Harris, Tenesa and Liewald2012; Plomin et al., Reference Plomin, Haworth, Meaburn, Price and Davis2013), personality (Vinkhuyzen et al., Reference Vinkhuyzen, Pedersen, Yang, Lee, Magnusson and Iacono2012), and economic and political preferences (Benjamin et al., Reference Benjamin, Cesarini, van der Loos, Dawes, Koellinger and Magnusson2012). However, the first GCTA report for children's behavior problems as rated by their parents, teachers, and themselves found no significant genetic influence (Trzaskowski, Dale, & Plomin, Reference Trzaskowski, Dale and Plomin2013).
Like early twin studies, GCTA has primarily been used to demonstrate heritability but, like twin studies, the technique can be used to go beyond the estimation of heritability. For example, GCTA has confirmed one of the most interesting and puzzling findings about cognitive development: the heritability of general cognitive ability increases during development (Trzaskowski, Yang, Visscher, & Plomin, Reference Trzaskowski, Yang, Visscher and Plomin2013). One interesting feature of GCTA is that, because it relies on comparisons between unrelated individuals, GCTA is entirely a between-family analysis, in contrast to traditional quantitative genetic methods like the twin method, which are within-family analyses, essentially comparing differences within pairs of twins in a family to differences between families in the population. For developmentalists interested in gene–environment interplay, this feature of GCTA is important because it opens up the possibility of investigating genetic influence on family-wide environmental measures rather than being constrained to investigating child-specific environmental factors that differ for children in a family.
However, GCTA estimates of genetic influence are generally only about half the heritabilities estimated by twin studies. Although this could be because twin studies have overestimated heritability, it is clear that GCTA underestimates heritability for two reasons (Yang et al., Reference Yang, Lee, Goddard and Visscher2011). First, GCTA only detects genetic effects that can be detected by the common SNPs (allele frequencies >1%) that happen to be incorporated in the DNA microarrays used in GWA studies. Second, GCTA is limited to detecting the additive effects of SNPs; it does not include gene–gene (or gene–environment) interaction. However, for these same reasons, GCTA provides critical clues for solving the missing heritability problem because GCTA gauges the extent to which the additive effects of common SNPs on current DNA arrays can potentially close the missing heritability gap if samples are sufficiently large. The answer is generally about 50%. The rest of the missing heritability might be found using rare DNA variants, other types of DNA variants, or nonadditive (gene–gene and gene–environment) interactions (Plomin, Reference Plomin2013). When whole-genome sequence is available for large samples of individuals, GCTA will be able to include all types of DNA variants.
The value of GCTA has been greatly increased by extending it beyond the univariate analysis of the variance of a single trait to the bivariate analysis of the covariance between two traits (Lee, Yang, Goddard, Visscher, & Wray, Reference Lee, Yang, Goddard, Visscher and Wray2012). The first application of this approach was to longitudinal IQ data from age 11 to age 70 (Deary et al., Reference Deary, Yang, Davies, Harris, Tenesa and Liewald2012), in which GCTA bivariate results showed that the remarkable phenotypic stability of IQ across 60 years from childhood to later life (phenotypic r = .63) is largely driven by genetic stability (genetic correlation = .62). Bivariate GCTA has also confirmed important twin study findings about cognitive development, such as the extensive overlap (pleiotropy) of genetic effects on diverse cognitive abilities (Plomin, Haworth, et al., Reference Plomin, Haworth, Meaburn, Price and Davis2013; Trzaskowski, Davis, et al., Reference Trzaskowski, Davis, DeFries, Yang, Visscher and Plomin2013; Trzaskowski, Shakeshaft, & Plomin, Reference Trzaskowski, Shakeshaft and Plomin2013). An important feature of bivariate GCTA analysis is that its estimates of genetic correlations are similar to twin-study estimates because GCTA estimates of genetic correlations are unbiased even though GCTA estimates of genetic variance and covariance underestimate twin-study estimates (Trzaskowski, Yang, et al., Reference Trzaskowski, Davis, DeFries, Yang, Visscher and Plomin2013). Extending GCTA from bivariate analysis to truly multivariate analysis will be especially valuable.
In the future, although traditional quantitative genetic designs, such as the twin design, will continue to make major contributions to developmental research, GCTA will provide a new approach that brings the potential of quantitative genetic analysis to any large sample of unrelated individuals for whom genomewide genotypes are available. The promise of GCTA will be more fully realized as it is used to go beyond simply confirming the heritability of traits to investigate developmental, multivariate, and gene–environment questions.
Despite the importance of having this completely new tool of GCTA in the armamentarium of quantitative genetics, nothing will advance the future of genomics research for developmentalists more than identifying the specific G, C, T, and A sequence variation responsible for heritability. In the next section, we briefly describe progress in this direction.
Finding the Missing Heritability
In summary, quantitative genetic research, which identifies genetic influence of any kind, finds substantial heritability for most traits and can go beyond the rudimentary question of heritability to address developmental, multivariate, and gene–environment issues. GCTA, which is currently limited to detecting additive effects of common variants on commercially available DNA arrays, predicts that such effects can account for about half of the heritability. In this section, we consider the implications for future attempts to identify the DNA variants responsible for heritability. It would be wonderful if we were able to say that genomic research has served up a menu of genes to be chosen by developmentalists for use in their research, but this is not yet the case. However, progress is being made, and it is important that developmental researchers be ready to take advantage of this genomics research, which will eventually make genomics relevant for all developmental researchers, as described later.
In this section, we focus on the implications of findings from GWA studies, which have dominated genomic research throughout the life sciences during the past 5 years. We will skip thousands of reports on linkage and candidate gene association. Linkage is systematic, using a few hundred DNA markers across the genome, to identify the chromosomal location of major gene effects by examining the cosegregation within family pedigrees between a DNA marker and a disorder. However, linkage is not powerful in detecting smaller genetic effects. Candidate gene association is powerful but not systematic. Association is a correlation in the population between an allele and a trait. For example, for late-onset Alzheimer disease, the frequency of a particular allele for the apolipoprotein E gene on chromosome 19 is about 40% in individuals with Alzheimer disease and about 15% in the rest of the population, one of the largest effect sizes found for a complex trait. The weakness of allelic association is that an association can only be detected if a DNA marker is itself the functional gene (called direct association) or very close to it (called indirect association or linkage disequilibrium). As a result, hundreds of thousands of DNA markers need to be genotyped to scan the genome thoroughly. For this reason, until very recently, allelic association has been used primarily to investigate associations with genes thought to be candidates for association.
A problem with the candidate gene approach is that we often do not have strong hypotheses as to which genes are candidate genes. Indeed, any of the thousands of genes expressed in the brain could be considered candidate genes for most behavioral traits. Moreover, candidate gene studies are limited to the 2% of the DNA that lies in coding regions. The biggest practical problem is that reports of candidate gene associations have been difficult to replicate (Tabor, Risch, & Myers, Reference Tabor, Risch and Myers2002), and they have not shown up in GWA studies (Siontis, Patsopoulos, & Ioannidis, Reference Siontis, Patsopoulos and Ioannidis2010). For example, a recent study of nearly 10,000 individuals was not able to replicate associations for 10 of the most frequently reported candidate gene associations for general cognitive ability (Chabris et al., Reference Chabris, Hebert, Benjamin, Beauchamp, Cesarini and van der Loos2012). Nonetheless, as discussed later, candidate gene studies will continue, especially as they move beyond a single SNP to gene-based and network-based analyses and as they are nominated as candidates on the basis of adequately powered empirical evidence.
In contrast, as explained elsewhere specifically for behavioral developmentalists (Plomin, Reference Plomin2013; Vrieze, Iacono, & McGue, Reference Vrieze, Iacono and McGue2012), GWA uses DNA arrays with hundreds of thousands of common SNPs that cover the entire genome in a systematic and atheoretical attempt to identify specific DNA variants additively associated with a trait. GWA has been successful in identifying hundreds of genes for hundreds of common diseases and quantitative traits (Sullivan, Reference Sullivan2012; Visscher, Brown, McCarthy, & Yang, Reference Visscher, Brown, McCarthy and Yang2012).
The largest effects are very small
Despite the success of GWA, an important lesson from these studies is that the largest effect sizes for the additive effects of common SNPs on current DNA arrays are astonishingly small in the population. Although there are thousands of single-gene disorders with huge effects on affected individuals, these disorders are rare and thus do not individually account for detectable variance in the population.
GWA results from individual studies have been combined in meta-analyses to reach the huge sample sizes needed to detect such small effects after correcting for multiple testing of hundreds of thousands of SNPs. GWA analyses with the largest sample sizes have been reported for the quantitative traits of height and weight. Weight has yielded the largest GWA effect size so far for a quantitative trait. One SNP in a gene known as fat mass and obesity associated protein accounts for almost 1% of the variance of weight in independent samples (Speliotes et al., Reference Speliotes, Willer, Berndt, Monda, Thorleifsson and Jackson2010). However, in GWA analyses of 250,000 individuals, the effect sizes for the next largest associations were closer to 0.1%, comparable to a correlation of .03, which is similar to the largest effect sizes found for height in a meta-analysis of 183,000 individuals (Lango Allen et al., Reference Lango Allen, Estrada, Lettre, Berndt, Weedon and Rivadeneira2010). A meta-analytic GWA study of structural MRI measures of volumes of brain regions for nearly 20,000 individuals found that the largest effect sizes accounted for 0.2% of the variance for hippocampal volume, 0.3% for intracranial volume, and 0.2% for total brain volume (Stein et al., Reference Stein, Medland, Vasquez, Hibar, Senstad and Winkler2012).
The largest effect sizes for behavioral traits are also tiny. For example, the largest study to date for a behavioral trait has recently been reported for the complex trait of total years of education (Rietveld et al., Reference Rietveld, Medland, Derringer, Yang, Esko and Martin2013). In a GWA meta-analysis of more than 100,000 individuals, three SNPs reached genomewide significance and were replicated in a sample of 25,000, but the largest association accounted for only 0.02% of the variance, that is, a correlation of .014. In a GWA meta-analysis of IQ for nearly 18,000 children, the largest effect size accounted for 0.2% of the variance, although even this association did not reach genomewide significance in the discovery sample (Benyamin et al., Reference Benyamin, Pourcain, Davis, Davies, Hansell and Brion2013).
Such small effect sizes are not limited to quantitative traits such as height, weight, and IQ. For example, large samples of cases have been amassed for psychiatric disorders. The largest effect size found for schizophrenia yielded an odds ratio of 1.1 in a GWA meta-analysis of more than 9,000 cases (Schizophrenia Psychiatric Genome-Wide Association Study [GWAS] Consortium, 2011). Similarly small effect sizes have been reported for the largest meta-analytic GWA studies of bipolar disorder with more than 7,000 cases (Psychiatric GWAS Consortium Bipolar Disorder Working Group, 2011), major depressive disorder with more than 9,000 cases (Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium, 2012), and attention-deficit/hyperactivity disorder (ADHD) with 3,000 cases (Neale et al., Reference Neale, Medland, Ripke, Asherson, Franke and Lesch2010). Associations for psychiatric disorders are smaller than those found for medical disorders, although even here the largest effect sizes involve odds ratios of less than 1.2 (Manolio, Reference Manolio2010; Visscher et al., Reference Visscher, Brown, McCarthy and Yang2012; Wellcome Trust Case Control Consortium, 2007).
Although the power of GWA to detect such tiny effects is limited, GWA studies have overwhelming power to prove that there are no large effects in the population. For example, a study of 20,000 individuals has 99.9% power to detect an association that accounts for 1% of the variance (i.e., a correlation of .10). GWA research implies that if the largest effects are so small, the smallest effects are likely to be infinitesimal. Incredibly large samples are needed to detect such small effects. For example, nearly 40,000 individuals are needed to detect an effect size of 0.1% (i.e., a correlation of .01) that reaches genomewide significance with 80% power.
Missing heritability from GWA research using common SNPs on current DNA arrays
Even though the effect sizes of individual SNPs are very small, their effects can be aggregated to estimate the size of the missing heritability gap. Genomewide significance may be too strict a criterion for selecting DNA variants from GWA studies because the conventional genomewide significance threshold of p < 5 × 10−8 requires samples of many hundreds of thousands to reach 80% power to detect such minuscule effect sizes. Selecting DNA variants using less stringent criteria generally explains more variance in independent samples (The International Schizophrenia Consortium, 2009). That is, although some false positive “associations” will be included, on balance, using relaxed criteria helps more than it hurts. For example, for weight, 32 replicated SNPs explain about 2% of the variance, but thousands of SNPs explain about 5% of the variance (Speliotes et al., Reference Speliotes, Willer, Berndt, Monda, Thorleifsson and Jackson2010). For height, 180 SNPs account for 10% of the variance, which increases to 13% when more SNPs are added (Lango Allen et al., Reference Lango Allen, Estrada, Lettre, Berndt, Weedon and Rivadeneira2010). For total years of education, the GWA meta-analysis mentioned earlier with more than 100,000 individuals explained about 1% of the variance using 3,500 SNPs and 2.5% of the variance using all 2.5 million SNPs (Rietveld et al., Reference Rietveld, Medland, Derringer, Yang, Esko and Martin2013). A similar pattern of results was reported for childhood IQ (Benyamin et al., Reference Benyamin, Pourcain, Davis, Davies, Hansell and Brion2013).
For medical disorders, where GWA consortia have been large enough to detect many reliable associations, specific SNPs in total account for up to about 8% of the liability (Lee et al., Reference Lee, Harold, Nyholt, Goddard, Zondervan and Williams2013). One recent example is coronary artery disease for which 150 SNPs accounted for about 5% of the total variance of liability (Deloukas et al., Reference Deloukas, Kanoni, Willenborg, Farrall, Assimes and Thompson2013). For schizophrenia, significant SNPs accounted for about 1% of the liability, and all SNPs accounted for almost 6% of the liability (Schizophrenia Psychiatric GWAS Consortium, 2011). For bipolar disorder, SNPs account for 1% to 3% of the liability (Psychiatric GWAS Consortium Bipolar Disorder Working Group, 2011).
Even in aggregate, the relatively small amount of variance explained by specific SNPs highlights the magnitude of the missing heritability problem. However, as indicated earlier, GCTA suggests that more heritability can be explained by additive effects of the common variants genotyped on current DNA arrays. As larger samples are amassed, the common SNPs on current DNA arrays will account for more of the missing heritability. The biggest question now in the genomic sciences is where the rest of the missing heritability can be found.
Beyond common SNPs on current DNA arrays
The most obvious direction for finding missing heritability is to consider less common DNA variants, including less common SNPs (Gibson, Reference Gibson2012). Common SNPs on currently available commercial DNA arrays have frequencies greater than 1% in the population. Many more SNPs are rarer, with frequencies that go down to “private mutations” unique to an individual. More than 10 million SNPs have been validated in populations around the world; only about 2 million have frequencies greater than 1% in the population studied.
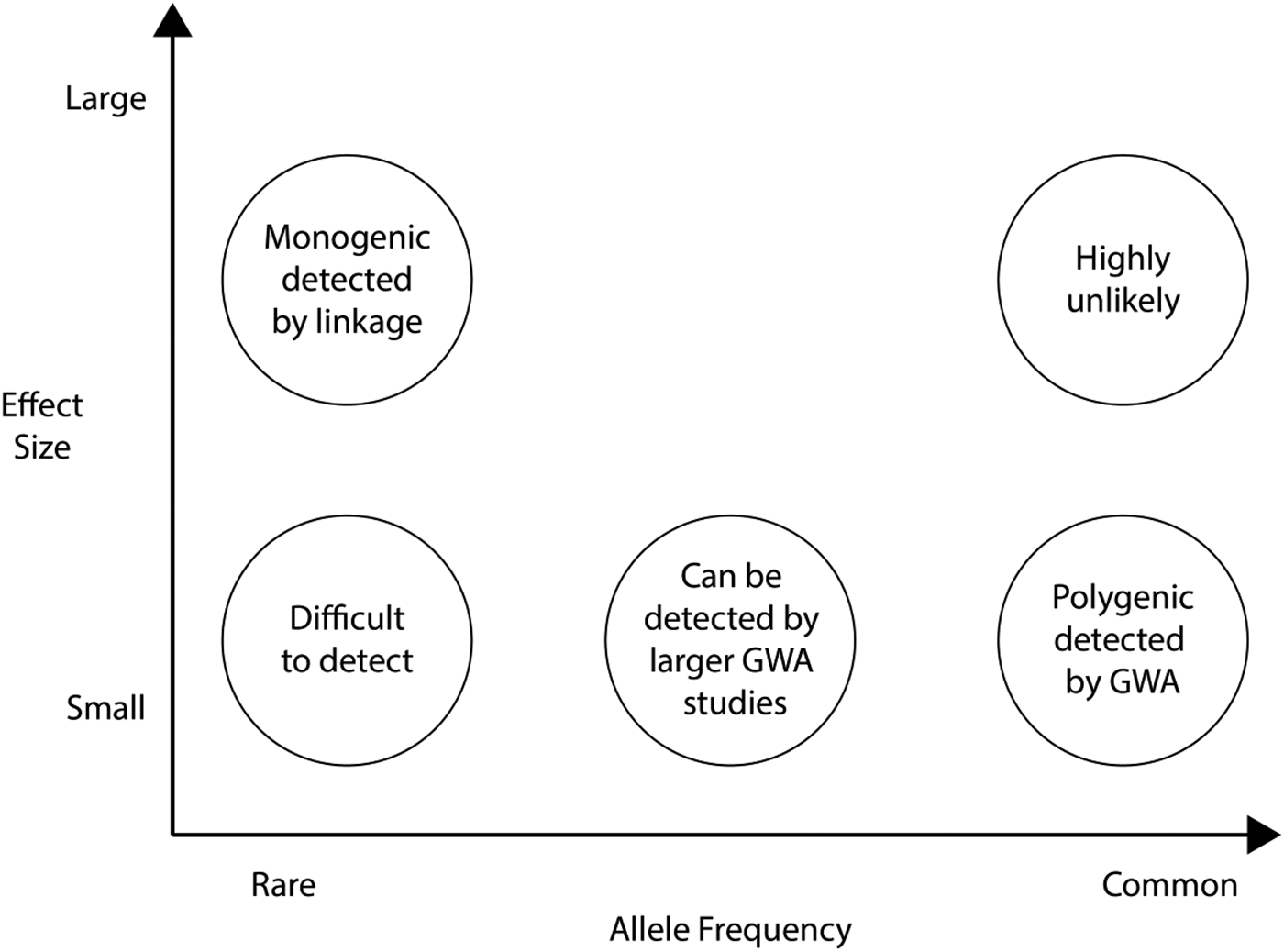
Figure 3 frames the issues by plotting allele frequency (from rare to common) against effect size (from small to large). Missing heritability could be found in all five circles in Figure 3, but the big question is the relative contributions of the five circles. So far, GWA has been limited to the lower right-hand corner (common SNPs of modest effect size); GCTA indicates that more of the missing heritability will be found here. As indicated in the upper left-hand corner, linkage analysis can detect rare alleles with large effects, which is why there has been a return to family-based designs (Ott, Kamatani, & Lathrop, Reference Ott, Kamatani and Lathrop2011). Associations in the upper right-hand corner (common alleles with large effect) are highly unlikely based on results so far. The most daunting prospect is the lower left-hand corner: very rare alleles of small effect, which will be extremely difficult to detect. In between these extremes, in the middle of Figure 3, is a promising area for finding some of the missing heritability: less common alleles (<1%) not well tagged by current microarrays yet common enough to show modest effects in the population.
Figure 3. Missing heritability: The relation between effect size and allele frequency. GWA, genomewide association. Adapted from “Genome-Wide Association Studies for Complex Traits: Consensus, Uncertainty and Challenges,” by M. I. McCarthy, G. R. Abecasis, L. R. Cardon, D. B. Goldstein, J. Little, J. P. A. Ioannidis, et al., Reference McCarthy, Abecasis, Cardon, Goldstein, Little and Ioannidis2008. Nature Reviews Genetics, 9, 356–369. Copyright 2008 by Nature Publishing Group. Adapted with permission.
The importance of rare variants has been highlighted by whole-exome sequencing, which takes the first step toward whole-genome sequencing by sequencing the 2% of DNA that is transcribed into RNA and then translated into amino acid sequences. An early finding is changing the traditional view of gene-disrupting mutations as rare and very damaging, even though that is the case for thousands of well-established monogenic diseases. It appears that each of us inherits about 100 rare loss of function variants that lead to about 20 genes that are completely inactivated but most of which have not been clearly associated with disease (MacArthur et al., Reference MacArthur, Balasubramanian, Frankish, Huang, Morris and Walter2012). Most of these are recessive alleles so that in the heterozygous state the other normal allele is able to do the gene's business, but even in the homozygous state, some of these gene-disrupting variants have no discernible phenotypic effect, suggesting a much more complicated system that tolerates gene disruption. Although these rare variants are not associated with known disorders, together they might well contribute to missing heritability even though individually their effects are small. A new DNA array called the Exome Chip includes most of the nonsynonymous SNPs in exons as rare as .001 (http://genome.sph.umich.edu/wiki/Exome_Chip_Design). The first paper using the Exome Chip has been published (Huyghe et al., Reference Huyghe, Jackson, Fogarty, Buchkovich, Stancakova and Stringham2013); many more such reports are in the pipeline. Although exomes are the location of most single-gene disorders, rare variants in the “dark matter” outside these traditional gene regions are also likely to contribute to heritability, as discussed below.
Even more surprising, whole-exome sequencing has also revealed that we each have several dozen noninherited (de novo) gene-disrupting mutations, some of which have been shown to contribute to sporadic cases of neurodevelopmental disorders, including intellectual disability, autism, and schizophrenia (Gratten, Visscher, Mowry, & Wray, Reference Gratten, Visscher, Mowry and Wray2013; Stein, Parikshak, & Geschwind, Reference Stein, Parikshak and Geschwind2013; Veltman & Brunner, Reference Veltman and Brunner2012). These de novo mutations may be more damaging than inherited mutations because they have not been subjected to generations of selective pressure. Again, although the effects of these rare mutations are small in the population, collectively they could contribute to missing heritability if the mutations occur in the germline. It is possible that common disorders might be collections of such rare variants in many genes that are different for different individuals (Mitchell, Reference Mitchell2012).
Before whole-exome sequencing uncovered so many single-nucleotide mutations, noninherited de novo variants were found that involved deletions and duplications of hundreds to millions of base pairs of DNA, called structural variants or copy number variants (CNVs; Conrad et al., Reference Conrad, Pinto, Redon, Feuk, Gokcumen and Zhang2010; Weischenfeldt, Symmons, Spitz, & Korbel, Reference Weischenfeldt, Symmons, Spitz and Korbel2013). Rare, large CNVs involving hundreds of thousands or even millions of base pairs have been shown to be risk factors for several common diseases (Wellcome Trust Case Control Consortium, 2010), including autism, ADHD, and schizophrenia (Rucker & McGuffin, Reference Rucker and McGuffin2012; Williams et al., Reference Williams, Franke, Mick, Anney, Freitag and Gill2012) and learning disabilities (Cooper et al., Reference Cooper, Coe, Girirajan, Rosenfeld, Vu and Baker2011; Gill, Reference Gill2012; Topper, Ober, & Das, Reference Topper, Ober and Das2011). The effects of such rare and large CNVs appear to be nonspecific, showing effects on multiple neurocognitive disorders (Malhotra & Sebat, Reference Malhotra and Sebat2012), and the effects are compounded for children with more than one such CNV (Girirajan et al., Reference Girirajan, Rosenfeld, Coe, Parikh, Friedman and Goldstein2012). Because many of these CNVs are not inherited, in that they appear in children but not their parents (i.e., de novo), they are especially sought in sporadic cases, that is, in cases whose families have no prior history of the disorder. Nonetheless, these CNVs can contribute to missing heritability because they can occur during the formation of gametes and thus will be shared by identical twins but are unlikely to be shared by fraternal twins. In addition, more than 10,000 smaller common CNVs have been identified with a frequency of at least 5% in the population (Conrad et al., Reference Conrad, Pinto, Redon, Feuk, Gokcumen and Zhang2010), although these are largely tagged by SNP arrays, which suggests that they will not make a major contribution to missing heritability (Wellcome Trust Case Control Consortium, 2010). We all have CNVs peppered throughout our genome without obvious effect, despite all of the extra or missing segments of DNA that include whole genes. However, these CNVs might have more subtle effects that contribute to the heritability of neurodevelopmental problems in the population (Geschwind, Reference Geschwind2011; Sahoo et al., Reference Sahoo, Theisen, Rosenfeld, Lamb, Ravnan and Schultz2011).
Variation in DNA sequence can be seen as a continuum from single nucleotide base pairs to small CNVs to larger CNVs to whole chromosomes. In addition, individuals differ in the number of certain repeating nucleotide base pairs, such as the triplet repeat CAG responsible for Huntington disease. Repeat sequence variants consist of two, three, or four base pairs that are repeated up to 100 times, creating multiple alleles, and have been found at as many as 50,000 loci throughout the genome. The value of whole-genome sequencing is that it will reveal variants of any kind.
Methods are also being developed to bring bioinformatics to bear on detecting small effects in the population rather than just relying on the blind brute force of larger samples. For instance, because a particular gene can have mutations at several locations, gene-based analyses can aggregate multiple DNA variants in a gene, including rare variants, which also reduces the multiple-testing problem (Bacanu, Reference Bacanu2012; Liu et al., Reference Liu, McRae, Nyholt, Medland, Wray and Brown2010). As an extreme example, hundreds of different mutations have been found in the gene responsible for phenylketonuria, and some of these different mutations have different effects (Scriver, Reference Scriver2007). At a broader level, it is possible to group genes in terms of pathway networks of related functions (Ramanan, Shen, Moore, & Saykin, Reference Ramanan, Shen, Moore and Saykin2012), including recent empirically based pathway discovery (Lehne & Schlitt, Reference Lehne and Schlitt2012). Another general example is to reduce the multiple-testing problem by focusing on DNA variants known to be functional, such as those known to affect gene expression (Pickrell et al., Reference Pickrell, Marioni, Pai, Degner, Engelhardt and Nkadori2010). Another example is to aggregate rare variants because many are likely to be deleterious (Li & Leal, Reference Li and Leal2008; Morris & Zeggini, Reference Morris and Zeggini2010).
These various strategies are likely to help find some of the missing heritability exposed by GWA studies relying on common SNPs. A worrying issue is the reliance of GWA research on additive effects. If heritability is substantially due to nonadditive gene–gene or gene–environment interactions, it will be extremely difficult to identify these effects. because power to detect two-way interactions is diminished (Vukcevic, Hechter, Spencer & Donnelly, Reference Vukcevic, Hechter, Spencer and Donnelly2011) and declines sharply with each additional interaction. We can only hope that quantitative genetic research is correct in its conclusion that most genetic variance is additive (Flint, DeFries, & Henderson, Reference Flint, DeFries and Henderson2004; Plomin, DeFries, et al., Reference Plomin, DeFries, Knopik and Neiderhiser2013). There are also evolutionary reasons to expect that most genetic variance is additive (Hill, Goddard, & Visscher, Reference Hill, Goddard and Visscher2008).
Much more attention should also be paid to better assessment of phenomes, not just genomes. The need for careful behavioral measurement, for including assessments of the environment, and for a developmental perspective are well covered in this Journal's Special Issue (McGrath, Weill, Robinson, Macrae, & Smoller, Reference McGrath, Weill, Robinson, Macrae and Smoller2012; Vrieze et al., Reference Vrieze, Iacono and McGue2012).
Polygenic Scores
How will this wealth of genomic data be incorporated in developmental research? As discussed above, the main message from GWA and GCTA research is that many DNA variants of small effect size are responsible for most of the heritability of complex traits and that DNA variants other than common SNPs will be part of the solution to the missing heritability problem. In the future, developmental research will use aggregate genotypic scores consisting of hundreds or thousands of DNA variants to predict children's genetic risk and resilience because these composite genotypic scores can be used with feasible sample sizes, even though huge samples are required to identify each association of small effect size. For example, if a polygenic predictor accounts for 5% of the variance (i.e., 10% of the heritable variance for a trait with 50% heritability), a sample size of 150 would have 80% power to detect its effect (p = .05, one tailed). These polygenic scores consisting of thousands of DNA variants will replace studies of a few SNPs in a candidate gene. With custom DNA arrays (see below), it will soon be possible to genotype hundreds of thousands of DNA variants for the current cost of genotyping a few candidate genes. Eventually, for a similar cost, it will be possible to sequence the entire genome (see below).
At least a dozen labels have been used to denote aggregated genotypic scores, such as polygenic susceptibility scores (Pharoah et al., Reference Pharoah, Antoniou, Bobrow, Zimmern, Easton and Ponder2002), genomic profiles (Khoury, Yang, Gwinn, Little, & Flanders, Reference Khoury, Yang, Gwinn, Little and Flanders2004), SNP sets (Harlaar et al., Reference Harlaar, Butcher, Meaburn, Sham, Craig and Plomin2005), and aggregate risk scores (The International Schizophrenia Consortium, 2009). Although most of these labels involve the notion of risk, we prefer the term polygenic score because it makes more sense for quantitative traits with positive as well as negative poles. This semantic distinction has some important implications. Some of the labels include SNP but the generic word polygenic is better because DNA variants other than SNPs, such as CNVs, will be included in aggregate genotypic scores. Unlike GCTA, which is a quantitative genetic technique for estimating genetic influence on average in a population without knowing the specific DNA variants responsible for heritability, polygenic scores are based on specific DNA variants associated with a trait.
DNA variants shown to be associated with a trait can simply be summed like items on a test. For example, 100 associations that each account for 0.1% of the variance of a trait on average would together account for 10% of the variance because DNA variants are uncorrelated unless they are very close together on a chromosome. Like positive and negative items on a questionnaire, the genotype scores for each DNA variant must be added in the correct direction. For SNPs, which have only two alleles (A1 and A2), there are three genotypes (A1A1, A1A2, A2A2) that can be assigned additive genotypic scores of 0, 1, and 2, respectively, to reflect increasing dosage of the A2 allele. This is an additive polygenic score, which is typically used for polygenic scores because heritability appears to be largely due to additive genetic factors, although nonadditivity at a locus (dominance) or between loci (epistasis) can also be incorporated in polygenic scores. Thus, if 100 SNPs were included in a polygenic score in this way, scores could vary from 0 to 200. Like a factor score for items on a test, when SNPs are summed to create a polygenic score, they are often weighted by the strength of their association with the phenotype (Dudbridge, Reference Dudbridge2013).
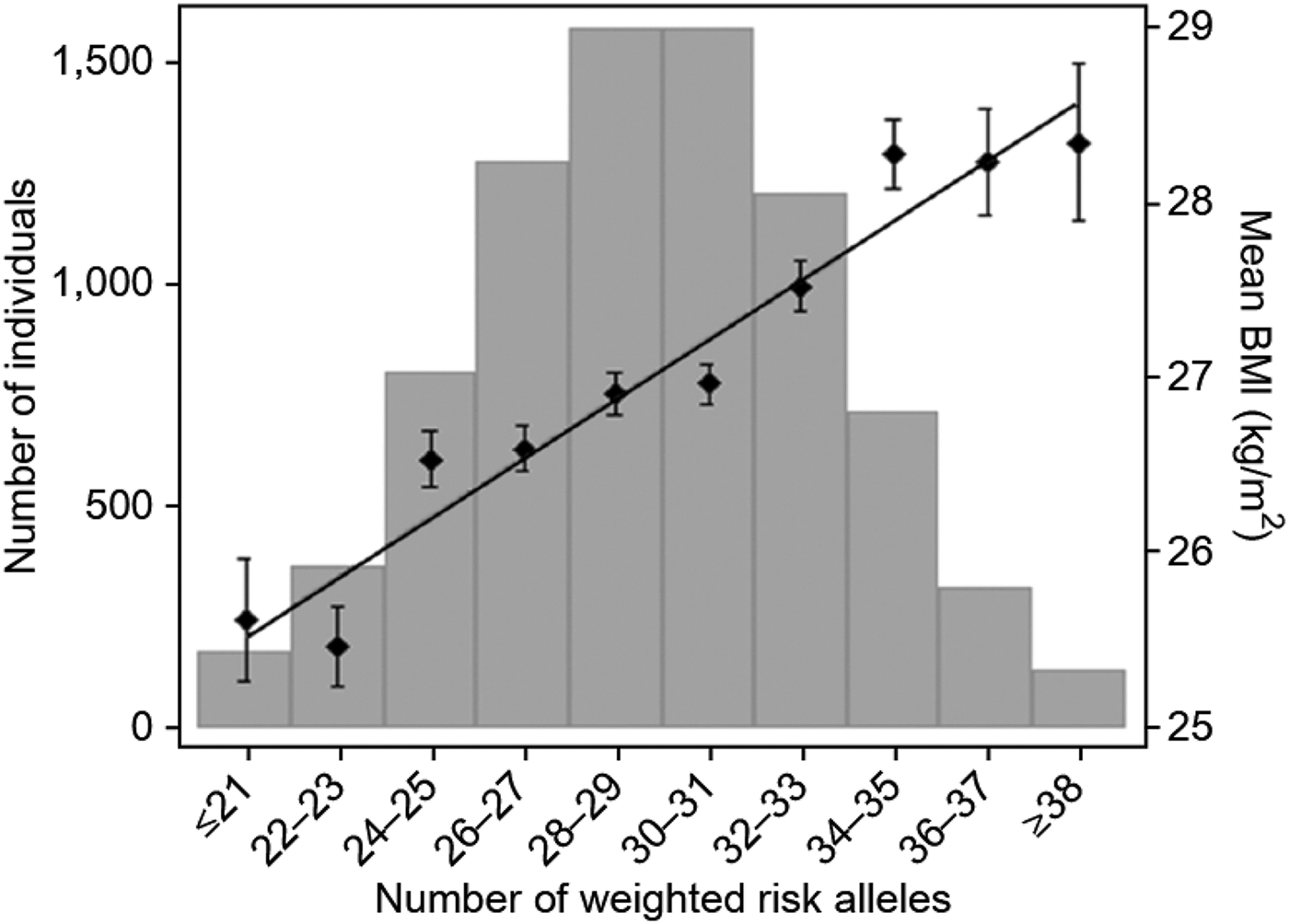
For example, as indicated earlier, a polygenic score for weight in adulthood created from 32 SNPs identified in GWA studies correlates .12 with weight in independent samples, which means that it accounts for 1.5% of the variance in weight or about 2% of the heritability of 70% (Speliotes et al., Reference Speliotes, Willer, Berndt, Monda, Thorleifsson and Jackson2010). Even with such a modest correlation, the top and bottom 2% of the weight distribution differ by about 8 kg (see Figure 4). This opens up the possibility of genotypic selection of individuals with low versus high polygenic scores from large samples for whom genomewide genotypes are available.
Figure 4. A polygenic score based on 32 single nucleotide polymorphisms associated with weight corrected for height (body mass index [BMI]) correlates 0.12 with BMI. Although the polygenic score only accounts for 1.5% of the total variance of BMI, individuals with the lowest and highest 2% polygenic scores differ by 8 kg, suggesting the possibility of reasonably sized research samples selected solely on the basis of polygenic scores (genotypic selection). Reprinted from “Association Analyses of 249,796 Individuals Reveal 18 New Loci Associated With Body Mass Index,” by E. K. Speliotes, C. J. Willer, S. I. Berndt, K. L. Monda, G. Thorleifsson, A. U. Jackson, et al., Reference Speliotes, Willer, Berndt, Monda, Thorleifsson and Jackson2010. Nature Genetics, 42, 940 (figure 2.1). Copyright 2010 by Nature Publishing Group. Reprinted with permission.
Polygenic scores can be used in the same way that candidate genes have been used. A neuroscientist might not find a polygenic score useful for investigating molecular pathways between genes and behavior through the brain, except perhaps to emphasize the need for a network approach governed by pleiotropy (each gene affects many traits) and polygenicity (each trait is affected by many genes). However, developmental researchers can use polygenic scores to investigate developmental, multivariate, and gene–environment issues as genetic predictors.
For example, a polygenic score constructed from SNPs associated with adult body mass index (BMI) has been shown to be just as strongly associated with BMI in childhood, suggesting substantial genetic stability for BMI between childhood and adulthood (Belsky et al., Reference Belsky, Moffitt, Houts, Bennett, Biddle and Blumenthal2012). Other interesting developmental results from this study include finding that the polygenic BMI score was associated with weight gain from birth to age 3 and with adiposity rebound at age 6 but not with birth weight.
Two conceptual implications of polygenic scores will affect future developmental research. First, the genetic liabilities for common disorders are normally distributed, which implies that from a genetic perspective there are no common disorders, just the extremes of quantitative liabilities. Second, polygenic scores will draw attention to the positive end of the normal distribution of polygenic scores, which has been called positive genetics (Plomin, Haworth, & Davis, Reference Plomin, Haworth and Davis2009). Using Figure 4 as an example, who are the children with low polygenic scores for obesity risk? Do they merely have the absence of risk factors or are there different mechanisms involved, such as being especially resistant to the temptations of a fast-food nation? The positive end of polygenic scores will stimulate research on how children flourish rather than fail and about resilience rather than vulnerability. However, it is also possible that being at very low risk for obesity has its own problems, such as being a finicky eater more prone to eating disorders. Perhaps the positive end of polygenic score distributions is not actually positive; “all things in moderation” might apply to polygenic scores as well.
In the near future, custom DNA arrays will become available that will assess hundreds of thousands of DNA variants relevant to particular traits at a cost less than the current cost of genotyping a few candidate genes. For example, custom arrays are available for several hundred thousand DNA variants related to immunology (ImmunoChip), cardiovascular functioning (CardioChip), and metabolic functioning (Metabochip). DNA arrays could be customized to produce polygenic scores for any and all aspects of behavioral development, including age-specific associations and associations involved in gene–environment interactions and correlations.
The strengths of DNA arrays include their speed, accuracy, and low cost. However, the problem with DNA arrays is that as new DNA variants relevant to a trait continue to be identified and need to be added to a polygenic score, it could be necessary to genotype the sample again using another DNA array. The huge advantage of whole-genome sequencing is that, once a genome is sequenced, there is no more genotyping to be done. A tipping point will come in the next few years as the plummeting cost of whole-genome sequencing approaches the cost of DNA arrays.
Pleiotropic Polygenic Scores
An important finding from quantitative genetics is that genetic effects tend to be general across phenotypes rather than specific to a single phenotype. In genetics, pleiotropy denotes the manifold effects of genes; that is, each gene affects many traits. Pleiotropy might drive polygenicity: if each gene affects many traits then many traits will be influenced by many genes. Pleiotropy has been highlighted in multivariate genetic research in the domain of cognitive and learning abilities and disabilities, where genetic correlations are as high as 0.80 between diverse cognitive processes, such as verbal and nonverbal cognitive abilities and reading and mathematics (Plomin & Kovas, Reference Plomin and Kovas2005), findings that have been confirmed using bivariate GCTA, as noted earlier. Substantial pleiotropy has also been found for childhood psychopathology (Lichtenstein, Carlstrom, Rastam, Gillberg, & Anckarsater, Reference Lichtenstein, Carlstrom, Rastam, Gillberg and Anckarsater2010) and adult psychosis (Lichtenstein et al., Reference Lichtenstein, Yip, Bjork, Pawitan, Cannon and Sullivan2009), which suggests a genetic architecture that differs greatly from current diagnostic classifications based on symptoms. For example, even though schizophrenia and bipolar disorder are the first branching point in psychiatric diagnosis, the genetic correlation between them is .60 (Lichtenstein et al., Reference Lichtenstein, Yip, Bjork, Pawitan, Cannon and Sullivan2009). GWA studies have come face to face with the reality of this quantitative genetic evidence for genetic overlap because most SNPs and CNVs associated with schizophrenia have turned out to be associated with bipolar disorder and vice versa (Girirajan et al., Reference Girirajan, Rosenfeld, Coe, Parikh, Friedman and Goldstein2012; Huang et al., Reference Huang, Perlis, Lee, Rush, Fava and Sachs2010; Liu et al., Reference Liu, Blackwood, Caesar, de Geus, Farmer and Ferreira2011; Malhotra & Sebat, Reference Malhotra and Sebat2012; Psychiatric GWAS Consortium Bipolar Disorder Working Group, 2011; Schizophrenia Psychiatric GWAS Consortium, 2011). The first polygenic score created for schizophrenia was found to be almost as highly correlated with liability for bipolar disorder (The International Schizophrenia Consortium, 2009). Extensive overlap of DNA variants has also been found for other medical disorders, especially autoimmune diseases (Cotsapas et al., Reference Cotsapas, Voight, Rossin, Lage, Neale and Wallace2011; Zhernakova et al., Reference Zhernakova, Stahl, Trynka, Raychaudhuri, Festen and Franke2011).
Rather than attempting to identify genes associated with one trait and then asking whether the genes are also associated with other traits, one direction for research is multivariate GWA studies that focus on genetic associations in common across traits. A multivariate GWA analysis of five major psychiatric disorders, including autism spectrum disorder and ADHD, found three SNPs that were associated with all five disorders in a meta-analysis of more than 30,000 cases (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013). This approach is multivariate in the sense that, although it begins with separate GWA analyses for each disorder, it looks for SNPs that are most associated across disorders regardless of whether the SNPs are among the most highly significant for any particular disorder. If an SNP is associated with two traits, it contributes to the phenotypic covariance and to the genetic correlation between the traits.
However, a more truly multivariate GWA analysis would focus on the covariance per se between traits rather than the variance of each trait. This is easier to see for quantitative traits: rather than beginning with univariate GWA analyses of each trait and then asking which SNPs show associations across traits, it would be more direct and more powerful to conduct GWA analyses of the covariance between traits. For example, two traits (X, Y) or more could be included in a multiple regression predicting SNP genotypes: the betas for X and Y would index trait-specific associations, and the multiple R would index the association due to covariance between X and Y. Software for such multivariate genetic analyses is available (O'Reilly et al., Reference O'Reilly, Hoggart, Pomyen, Calboli, Elliott and Jarvelin2012).
Finding DNA variation that accounts for this pervasive pleiotropy could have far-reaching implications for assessing and understanding typical and atypical development. Pleiotropic polygenic scores could be created that predict what is in common across broad domains such as psychopathology and cognitive abilities and disabilities. These pleiotropic polygenic scores could be used to look at the DNA-based architecture of psychopathology and cognition free from a century of phenotypic diagnoses and definitions. Future research can explore the psychopathological and cognitive traits that best characterize these pleiotropic polygenic factors and the mechanisms that underlie them, chart their developmental course, and consider their interplay with the environment. Trait-specific polygenic scores that remove the considerable genetic variance spread across traits are also likely to provide cleaner targets for research on typical and atypical development.
The End of Genotyping: Whole-Genome Sequencing
Rather than genotyping particular DNA variants on a DNA array, whole-genome sequencing has two huge advantages: it reveals all variants in the entire DNA sequence and it only needs to be done once. The cost of whole-genome sequencing is dropping so quickly that it will soon become competitive with the cost of DNA arrays. Before DNA arrays became available, it cost about $1 to genotype one SNP for one individual; in other words, genotyping a million SNPs for just one individual would cost $1 million, which is why GWA studies were not feasible other than pooling DNA for low and high groups (e.g., Plomin et al., Reference Plomin, Hill, Craig, McGuffin, Purcell and Sham2001). When commercially available DNA arrays became available in the early 2000s, it cost about $1,000 for a DNA array that genotyped 10,000 SNPs for one individual (i.e., $0.10 per genotype), but in a couple of years it cost less for a DNA array that genotyped 100,000 SNPs (<$0.01 per genotype). Now it costs about $500 for an array that genotypes a million SNPs (~$0.0001 per genotype). Despite the low cost per genotype, the average GWA sample size of 2,000 will cost $1 million, and, as discussed earlier, we now know that a sample size of 2,000 is much too small to detect the expected small effect sizes.
The cost of sequencing the entire genome of 3 billion nucleotide base pairs of DNA has also plummeted. The first human genome sequence was announced in 2003 and took 2,000 researchers 5 years at a cost of approximately $2 billion (http://www.genome.gov/11006943). Sequencing the entire genome of an individual now takes 1 day and costs about $5,000. A completely new technology is predicted to bring the cost down to under $1000 (Maitra, Kim, & Dunbar, Reference Maitra, Kim and Dunbar2012).
Because sequencing yields all DNA sequence variation, it means that no more genotyping is required. This has far-reaching implications for developmentalists: if an individual's DNA sequence is known, there is no need to do any further genotyping for and no need to obtain DNA from that individual. Although storage, quality control, and analysis of such massive amounts of data is currently challenging (Altmann et al., Reference Altmann, Weber, Bader, Preuss, Binder and Muller-Myhsok2012), as whole genome sequencing becomes routine, so too will the necessary data management and analytic tools, as has happened for analysis of DNA array data.
As the cost of sequencing continues to fall, many individuals will pay to have their DNA sequenced, just as hundreds of thousands of people have already paid to have their DNA genotyped on DNA arrays by direct to consumer companies, such as 23&Me, to detect 250 single-gene diseases and determine ancestry (https://www.23andme.com/). There are, of course, many concerns with direct to consumer genotyping (Guttmacher, McGuire, Ponder, & Stefansson, Reference Guttmacher, McGuire, Ponder and Stefansson2010), and these concerns will be greatly magnified with direct to consumer sequencing, which is already on offer. Parents have also begun to pay to have their children's DNA sequenced (Rochman, Reference Rochman2012). Indeed, it is likely that in some countries, all children will have their DNA sequenced. In an excellent book on the potential of the genomics revolution for personalized medicine, Francis Collins, former director of the Human Genome Project and currently director of the US National Institutes of Health, after noting some caveats and cautions, makes this prediction: “I am almost certain that complete genome sequencing will become part of newborn screening in the next few years . . . It is likely that within a few decades people will look back on our current circumstance with a sense of disbelief that we screened for so few conditions” (Collins, Reference Collins2010, p. 50).
If this prediction is correct, it will be a game-changer for developmentalists. If DNA sequence data were widely available for children, whether through direct to consumer testing or through systematic neonatal screening, developmentalists would no longer need to collect DNA, to genotype it, or to sequence it. Our long-term prediction for the future of genomics for developmentalists is the routine use of polygenic scores derived from DNA sequence data. Developmental clinicians can use polygenic scores to provide diagnostic information based on etiology rather than symptomatology, suggest personalized interventions, and enable early prediction that will lead to prevention. Developmental researchers can use polygenic scores to predict genetic propensities for children and to trace how those genetic dispositions develop longitudinally, how they overlap with other traits, and how they interact and correlate with the environment. Polygenic scores associated with behavioral traits will lead to research attempting to understand links between the genome, epigenome, transcriptome, proteome, neurome, and behavior. The grandest implication for science is that genomics will serve as a common denominator integrating all the life sciences.
Glossary
additive genetic variance Individual differences caused by the independent effects of alleles or loci that “add up,” in contrast to nonadditive genetic variance, in which the effects of alleles or loci interact.
allele An alternative form of a gene at a locus, for example, A1 versus A2
allelic association An association between allelic frequencies and a phenotype. For example, the frequency of allele 4 of the gene that encodes apolipoprotein E is about 40% for individuals with Alzheimer disease and 15% for control individuals who do not have the disorder.
allelic frequency Population frequency of an alternate form of a gene. For example, the frequency of the PKU allele is about 1%.
base pair One step in the spiral staircase of the double helix of DNA, consisting of adenine bonded to thymine or cytosine bonded to guanine
bioinformatics Techniques and resources to study the genome, transcriptome, and proteome, such as DNA sequences and functions, gene expression maps, and protein structures
candidate gene A gene whose function suggests that it might be associated with a trait. For example, dopamine genes are considered as candidate genes for hyperactivity because the drug most commonly used to treat hyperactivity, methylphenidate, acts on the dopamine system.
coding region The portion of a gene's DNA composed of exons that code for proteins
copy number variant (CNV) A structural variation that involves duplication or deletion of long stretches of DNA (1000 to many 1000s of base pairs in length), often encompassing protein-coding genes as well as noncoding genes. CNVs account for more than 10% of the human genome.
direct association An association between a trait and a DNA marker that is the functional polymorphism that causes the association (in contrast to indirect association, in which the DNA marker is not the functional polymorphism)
DNA The double-stranded molecule that encodes genetic information. The two strands are held together by hydrogen bonds between two of the four bases, with adenine bonded to thymine and cytosine bonded to guanine.
DNA array A collection of DNA probes (short DNA sequences) attached to a solid surface used to genotype hundreds of thousands of DNA sequence variants, also called DNA microarray or DNA chip
DNA marker A polymorphism in DNA itself, such as a SNP or CNV
DNA sequence The order of base pairs on a single chain of the DNA double helix
dominance The effect of one allele depends on that of another. A dominant allele produces the same phenotype in an individual regardless of whether one or two copies are present. (Compare with epistasis, which refers to nonadditive effects between genes at different loci.)
effect size The proportion of individual differences for the trait in the population accounted for by a particular factor. For example, heritability estimates the effect size of genetic differences among individuals.
endophenotype An “inside,” or intermediate, phenotype that does not involve overt behavior
environome All environmental factors that affect a phenotype
epigenetics DNA methylation or histone modifications that affect gene expression without changing DNA sequence, can be involved in long-term developmental changes in gene expression
epigenome Epigenetic events throughout the genome
epistasis Nonadditive interaction between genes at different loci. The effect of one gene depends on that of another. (Compare with dominance, which refers to nonadditive effects between alleles at the same locus.)
exome All exons in the genome
exon The 2% of DNA transcribed into messenger RNA and translated into protein
gamete Mature reproductive cell (sperm or ovum) that contains a haploid (half) set of chromosomes
gene The basic unit of heredity, a sequence of DNA bases that codes for a particular product, includes DNA sequences that regulate transcription (see also allele; locus)
gene expression Transcription of DNA into mRNA
genetics Methods and theory related to inheritance. Includes both quantitative genetic and molecular genetic approaches
genome All the DNA sequences of an organism. The human genome contains about 3 billion DNA base pairs across 23 pairs of chromosomes
genomewide association An association (correlation) between DNA sequence variation and a phenotype found in a systematic search throughout the genome
genomewide complex trait analysis (GCTA) A technique to estimate the extent to which phenotypic variance for a trait can potentially be explained by all genotyped DNA variants. For a sample of thousands of individuals, overall genotypic similarity pair by pair is used to predict phenotypic similarity. Does not identify specific allelic associations.
genomics Theory and methods that investigate DNA sequence variation throughout the genome
genotype An individual's combination of alleles at a particular locus
heritability The proportion of phenotypic differences among individuals that can be attributed to genetic differences in a particular population. Broad-sense heritability involves all additive and nonadditive sources of genetic variance, whereas narrow-sense heritability is limited to additive genetic variance.
heterozygosity The presence of different alleles at a given locus on both members of a chromosome pair
homozygosity The presence of the same allele at a given locus on both members of a chromosome pair
indirect association An association between a trait and a DNA marker that is not itself the functional polymorphism that causes the association. In contrast to direct association, in which the DNA marker itself is the functional polymorphism.
linkage Loci that are close together on a chromosome. Linkage is an exception to Mendel's second law of independent assortment, because closely linked loci are not inherited independently within families.
linkage disequilibrium A violation of Mendel's law of independent assortment, in which genes are uncorrelated. It is most frequently used to describe how close DNA markers are together on a chromosome; linkage disequilibrium of 1.0 means that the alleles of the DNA markers are perfectly correlated; 0.0 means that there is complete nonrandom association (linkage equilibrium).
locus (plural, loci) The site of a specific gene on a chromosome, Latin for “place”
methylation An epigenetic process by which gene expression is inactivated by adding a methyl group to a chromosome region
missing heritability The difference between the total variance accounted for by known genomewide associations and heritability estimates from quantitative genetic studies
molecular genetics The investigation of the effects of specific genes at the DNA level. In contrast to quantitative genetics, which partitions phenotypic variances and covariances into genetic and environmental components
multivariate genetic analysis Quantitative genetic analysis of the covariance between traits
nonadditive genetic variance Individual differences due to nonlinear interactions between alleles at the same (dominance) or different (epistasis) loci. (In contrast, see additive genetic variance.)
noncoding RNA (ncRNA) DNA that is transcribed into RNA but not translated into amino acid sequences (e.g., introns and microRNA)
nonsynonymous SNP A SNP that alters the amino acid sequence of a protein and is thus likely to make a functional difference. In contrast, synonymous SNPs do not alter the amino acid sequence of proteins.
odds ratio An effect size statistic for association calculated as the odds of an allele in cases divided by the odds of the allele in controls. An odds ratio of 1.0 means that there is no difference in allele frequency between cases and controls.
phenotype An observed characteristic of an individual that results from the combined effects of genotype and environment
pleiotropy Multiple effects of a gene
polygenic trait A trait influenced by many genes
polymorphism A locus with two or more alleles, Greek for “multiple forms”
proteome All the proteins translated from RNA (transcriptome)
quantitative genetics Theory and methods (such as the twin and GCTA methods for human analysis) to estimate overall genetic and environmental contributions to phenotypic variance and covariance in a population
repeat sequence variant Short sequences of DNA (2, 3, or 4 nucleotide bases of DNA) that repeat a few times to a few dozen times, used as DNA markers
single-nucleotide polymorphism (SNP) The most common type of DNA polymorphism, which involves a mutation in a single nucleotide. SNPs (pronounced “snips”) can produce a change in an amino acid sequence (called nonsynonymous, i.e., not synonymous).
transcription The synthesis of an RNA molecule from DNA in the cell nucleus
transcriptome RNA transcribed from all the DNA in the genome
translation Assembly of amino acids into peptide chains on the basis of information encoded in messenger RNA, occurs on ribosomes in the cell cytoplasm
whole-exome sequence Determining the complete sequence of DNA nucleotide base pairs for all exons in the genome
whole-genome sequencing Determining the complete sequence of DNA nucleotide base pairs for a genome