


Since the seminal Special Issue of Development and Psychopathology in 1994 (Cicchetti & Tucker, Reference Cicchetti and Tucker1994), major advances have taken place in research into brain plasticity and critical periods of development as they inform neuropsychiatric disorders. In this paper, we review the current concept of neuroplasticity as well as the expanding evidence of its aberrations in major psychiatric disorders, with a special focus on schizophrenia and the evolution of risk for this illness. We also examine the potential translational applications of our understanding of neuroplasticity, and how we may harness this in the service of treatment and prevention of serious mental disorders.
Historical Overview and Definitions
The great neurologists of the 19th century, including Ramon Cajal, the father of modern neuroscience, thought that once developed, the adult brain is unlikely to change with experience (Cajal, Reference Cajal1894). However, Cajal later suggested that memories might be formed by strengthening the connections between existing neurons (Stahnisch & Nitsch, Reference Stahnisch and Nitsch2002). Hughlings Jackson, the father of modern neurology, proposed the hierarchical nature of how the nervous system is organized, and he made the distinction between negative symptoms that result from loss of nervous function and positive symptoms that may represent a failed attempt to compensate for such functional loss by disinhibited activity of lower brain regions (Berrios, Reference Berrios2001). William James, the noted American psychologist, who was inspired by Jackson's work, was among the first to suggest that the brain is not as immutable as previously thought. In his book The Principles of Psychology, James wrote, “Organic matter, especially nervous tissue, seems endowed with a very extraordinary degree of plasticity” (James, Reference James1890). This view was ignored for several decades. Donald Hebb (Reference Hebb1949) proposed an idea later referred to as “Hebbian learning,” that is, when two neurons repeatedly or persistently fire together, some change takes place in one or both cells such that the efficiency of neuronal activity is increased. This adage that “neurons that fire together wire together” became the cornerstone of the concept of neuroplasticity, which refers to how the brain changes (organizes and reorganizes) in response to experience. While the brain shows plasticity throughout an individual's lifetime, its capacity for change may be higher at certain times than others; this led to the concept of critical periods.
Brain plasticity has been defined in a number of different ways (Figure 1). Plasticity can encompass both synaptic plasticity and nonsynaptic plasticity. Synaptic plasticity is the ability of a synapse between two neurons to change in strength over time, perhaps due to modifications in synaptic potentials or receptors that transmit chemical signals. Modification of synaptic strength is mediated by long-term potentiation (LTP), a phenomenon whereby repeated signal transmission between two neurons leads to long-lasting enhancement (Lomo, Reference Lomo2003). By contrast, nonsynaptic plasticity is a modification of the intrinsic excitability of the neuron, mediated through changes in structures such as the soma, the axon, or the dendrites. This may occur through neuronal transmission that happens outside of synapses (e.g., by extracellular diffusion processes), using processes such as volume transmission (Vizi, Reference Vizi1979) or via glial and vascular changes (Markham & Greenough, Reference Markham and Greenough2004).

Figure 1. (Color online) Determinants, mechanisms, and consequences of brain plasticity.
Neuroplasticity may occur in at least two (not mutually exclusive) developmental contexts (Figure 2). Very early in development, experience and its resulting neuronal activity can shape neuronal response properties regardless of an organism's attention to a stimulus. This process of experience-expectant neuroplasticity (Hubel & Wiesel, Reference Hubel and Wiesel1959) shapes neural representations to reflect statistical regularities in inputs (e.g., from one eye vs. another, and in the environment). Such plasticity is often conceptualized to occur within a finite window, an early “critical period.” Maladaptive experiences or insults to the developing brain during these critical periods can have lasting behavioral consequences. By contrast, experience-dependent neuroplasticity (Klintsova & Greenough, Reference Klintsova and Greenough1999) occurs throughout development. This process involves changes in neuronal activity in relation to experience, leading to lasting neural representations.
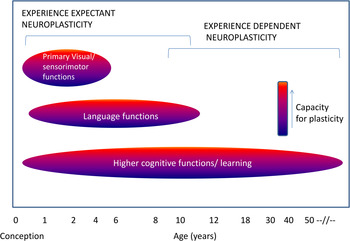
Figure 2. (Color online) Critical windows of neuroplasticity.
Based on the nature of experience and the state of the organism, the brain can be reshaped in either adaptive or maladaptive ways. Aberrant plasticity can have a profound impact on neuronal activity (Papa, De Luca, Petta, Alberghina, & Cirillo, Reference Papa, De Luca, Petta, Alberghina and Cirillo2014; Pirttimaki & Parri, Reference Pirttimaki and Parri2013) and may be triggered in pathological conditions such as Alzheimer disease and Huntington disease (Oberman & Pascual-Leone, Reference Oberman and Pascual-Leone2013). Maladaptive excessive plasticity has also been implicated in addiction, posttraumatic stress disorder, and depression (Pittenger, Reference Pittenger2013). As we will argue in this paper, the cardinal features of schizophrenia may arise from either diminished plasticity or pathologically excessive plasticity. Through the lens of premorbid and prodromal risk states for schizophrenia, and in an extension of Jackson's model, we propose that failure of plasticity in key brain circuits may result in cognitive and deficit symptoms, while an aberrant hyperplastic response to such deficits might underlie psychosis and emotional dysregulation (Figure 3).

Figure 3. (Color online) Schematic model representing possible ways in which plasticity processes may be impaired in schizophrenia.
Neurobiological Processes Underlying Plasticity
The nervous system is variably plastic throughout the life span. In this section, we will review the mechanisms of neuroplasticity as they relate to neurogenesis and apoptosis, synaptic formation and pruning, synaptic modulation, nonsynaptic processes, and neuronal support cells.
Neurogenesis
The earliest stages of nervous system development include generation of neurons and glial cells from stem cell progenitors, neural differentiation, and neuronal migration to other locations. After neuronal migration, growth of axons and dendrites occurs through extension of growth cones at their tips. This process is influenced by cell–cell adhesion molecules and other external molecular signals (Alberts et al., Reference Alberts, Johnson, Lewis, Raff, Roberts and Walter2002). Next, neurons compete for access to neurotrophic factors, and about half of them die through programmed cell death, also called apoptosis (Alberts et al., Reference Alberts, Johnson, Lewis, Raff, Roberts and Walter2002). Synapses begin to form between neurons, mediated by the release of neurotrophic factors by target tissues. In addition, neurogenesis continues during adulthood in the dentate gyrus of the hippocampus and the subventricular zone (Benarroch, Reference Benarroch2013; Lledo, Alonso, & Grubb, Reference Lledo, Alonso and Grubb2006).
Synaptic plasticity and role of glutamatergic neurotransmission
Synaptic plasticity is the capacity of synapses to change their strength in response to changes in their activity. LTP, a key mechanism underlying synaptic plasticity, is the strengthening of the transmission across two neurons with repeated stimulation of a synapse, reflected in changes to the amplitude of the postsynaptic potential (Zhang & Linden, Reference Zhang and Linden2003). LTP underlies memory formation and learning and occurs in many brain regions, notably the hippocampus. In early phase LTP, which occurs in the first several hours, large quantities of calcium ions are released and protein kinases are activated (Bliss & Collingridge, Reference Bliss and Collingridge1993). In late phase LTP, gene transcription occurs, and proteins are synthesized over the course of hours to days (Lynch, Reference Lynch2004). Brain-derived neurotrophic factor (BDNF) plays an important role in this phase. The molecular mechanisms underlying LTP include activation of N-methyl d-aspartate (NMDA) receptors, which serve as coincidence detectors when two neurons fire simultaneously, allowing flow of ions into the neuron. NMDA antagonists block LTP and learning (Morris, Anderson, Lynch, & Baudry, Reference Morris, Anderson, Lynch and Baudry1986).
Long-term depression (LTD) refers to a long-lasting decrease in synaptic strength. Like LTP, it also involves glutamate signaling on NMDA and AMPA receptors (Collingridge, Peneau, Howland, & Wang, Reference Collingridge, Peineau, Howland and Wang2010). In contrast to LTP, it is induced by long-lasting low-frequency stimulation, rather than brief high-frequency stimulation (Collingridge et al., Reference Collingridge, Peineau, Howland and Wang2010). Mechanisms of LTD may include reductions in glutamate release due to both presynaptic and postsynaptic changes, removal of AMPA receptors from the synapse, or changes in the conductance properties of receptors (Collingridge et al., Reference Collingridge, Peineau, Howland and Wang2010; Malenka, Reference Malenka2003). LTD, like LTP, may also be involved in neuropsychiatric disease. Stress enhances LTD in the hippocampus through activation of NMDA receptors (Kim, Foy, & Thompson, Reference Kim, Foy and Thompson1996), which may help explain stress-related impairments in memory formation. In addition, LTD has been hypothesized to be involved in synaptic refinement processes during development (Collingridge et al., Reference Collingridge, Peineau, Howland and Wang2010). Thus, disruptions of LTD may lead to aberrant plasticity during development.
Nonsynaptic plasticity and role of glia
Nonsynaptic plasticity includes a wide range of processes affecting the intrinsic excitability of neurons (Daoudal & Debanne, Reference Daoudal and Debanne2003). Mechanisms may include changes to the neuronal soma (cell body), dendrites, axons, and components of the neuronal membrane, such as resting and voltage-gated ion channels (Mozzachiodi, Lorenzetti, Baxter, & Byrne, Reference Mozzachiodi, Lorenzetti, Baxter and Byrne2008). Nonsynaptic plasticity may impact serotonin, acetylcholine, metabotropic glutamate, kainite, and NMDA receptors, voltage-gated calcium channels, and cellular signaling (Daoudal & Debanne, Reference Daoudal and Debanne2003; Zhang & Linden, Reference Zhang and Linden2003). Volume transmission, another aspect of nonsynaptic plasticity, involves both activation of extrasynaptic receptors and induction of activity by diffusion of molecules from the extracellular fluid into synaptic clefts. The role of nonsynaptic plasticity in memory and learning is still unclear.
Glial cells are nonneuronal cells that help maintain and support neurons, providing structural support and insulation, among other functions. While glial cells were generally considered as “support cells,” glia can dynamically respond to environmental input and influence neuronal function by releasing neurotransmitters (gliotransmitters). Astrocytes, a type of glial cell, wrap their membranous projections around synapses and release substances such as neurotransmitters (Paixao & Klein, Reference Paixao and Klein2010), and d-serine, which influences LTP and LTD (Henneberger, Papouin, Oilet, & Rusakov, Reference Henneberger, Papouin, Oliet and Rusakov2010). Glutamate transporters on astrocytes remove excess glutamate from the extracellular space, preventing the excitotoxicity that can result from excessive glutamate stimulation of the synapse (Rothstein, Reference Rothstein1996). Genetic or molecular changes that alter glutamate transporters in glia result in impairment of LTP (Filosa et al., Reference Filosa, Paixao, Honsek, Carmona, Becker and Feddersen2009) and LTD (Omrani et al., Reference Omrani, Melone, Bellesi, Safiulina, Aida and Tanaka2009).
Neurotrophins and other trophic proteins
Neurotrophins are signaling proteins that prompt neurons to grow and differentiate, and thus they are essential to neurodevelopment and neural plasticity. Several major neurotrophins have been studied in depth: BDNF, nerve growth factor, neurotrophin-3, and neurotrophin-4. The most investigated neurotrophin, BDNF, influences synaptic regulation and growth (Kleim et al., Reference Kleim, Chan, Pringle, Schallert, Procaccio and Jimenez2006) and neuronal migration and differentiation (Huang et al., Reference Huang, Kirkwood, Pizzorusso, Porciatti, Morales and Bear1999). BDNF is involved in late-phase LTP (Tartaglia et al., Reference Tartaglia, Du, Tyler, Neale, Pozzo-Miller and Lu2001) and may work partly by enhancing the response of synapses to tetanic stimulation (Figurov, Pozzo-Miller, Olafsson, Wang, & Lu, Reference Figurov, Pozzo-Miller, Olafsson, Wang and Lu1996).
Sleep, electrophysiology, and neuroplasticity
An important mediator of synaptic plasticity is sleep. According to the sleep homeostasis hypothesis, the extensive learning experiences and synaptic strengthening that occur during wakeful states results in synaptic fatigue at a cellular level, which is restored during sleep (Tononi & Cirelli, Reference Tononi and Cirelli2014). It is interesting that sleep spindles are thought to play a role in synaptic changes and sleep-dependent memory consolidation (Fogel et al., Reference Fogel, Martin, Lafortune, Barakat, Debas and Laventure2012). Spindles are nonrapid eye movement sleep EEG rhythms (7–14 Hz). Spindle-associated spike discharges have been shown to induce LTP-like synaptic plasticity, thus playing an important role in sleep-dependent memory consolidation (Rosanova & Ulrich, Reference Rosanova and Ulrich2005). Moreover, a simultaneous EEG functional magnetic resonance imaging (fMRI) study showed that the functional connectivity of the hippocampal formation with the neocortex was the strongest during Stage 2 nonrapid eye movement sleep when spindles were present (Andrade et al., Reference Andrade, Spoormaker, Dresler, Wehrle, Holsboer and Samann2011).
Electrophysiology has been used to assess LTP in the human cortex in the waking state as well. In normal individuals, repetitive auditory stimulation or visual stimulation has been associated with increases in the amplitude of the auditory and visual evoked potentials, respectively (Clapp, Hamm, Kirk, & Teyler, Reference Clapp, Hamm, Kirk and Teyler2012; Clapp, Kirk, Hamm, Shepherd, & Teyler, Reference Clapp, Kirk, Hamm, Shepherd and Teyler2005), suggesting the induction of LTP. This type of LTP generally lasts more than an hour and can be blocked by NMDA receptors in animal models (Clapp et al., Reference Clapp, Hamm, Kirk and Teyler2012). As will be discussed in detail later, the combination of transcranial magnetic stimulation and EEG is now being used to identify deficits of LTP in neuropsychiatric disease.
Network plasticity
The existence of cortical network plasticity is supported by functional neuroimaging studies of cortical remapping during learning. When a specific motor task is practiced repeatedly, the amount of motor cortex activated during performance of that task widens in comparison with performance of different, unpracticed tasks (Karni et al., Reference Karni, Meyer, Jezzard, Adams, Turner and Ungerleider1995). However, it is not known whether this cortical remapping of a learned task is permanent or a temporary part of the learning process. A recent expansion–normalization model (Kilgard, Reference Kilgard2012) suggests that changes in cortical mapping during learning are transient states that facilitate the learning process. Through a temporary increase in the availability of neurons to engage in a novel task, the optimal neural circuitry for the task can then be recruited and refined (Kilgard, Reference Kilgard2012).
Studies of brain injury also demonstrate the brain's adaptive cortical plasticity. Following a stroke, brain regions adjacent to the injured region are recruited to perform the tasks formerly performed by the injured region (Xerri, Merzenich, Peterson, & Jenkins, Reference Xerri, Merzenich, Peterson and Jenkins1998). Active rehabilitation training may enhance this process of cortical reorganization (Nudo & Milliken, Reference Nudo and Milliken1996). Over time, this task-related activation decreases and becomes restricted to fewer regions, implying an initial compensatory expansion of activation, followed by cortical reorganization (Ward, Brown, Thompson, & Frackowiak, Reference Ward, Brown, Thompson and Frackowiak2003).
Genes, environment, and plasticity
Genetic variation can influence plasticity processes, including neurogenesis and LTP. For example, mutations in the disrupted in schizophrenia 1 (DISC1) gene, which is associated with schizophrenia, can disrupt hippocampal neurogenesis, leading to creation of neurons with abnormal morphology or premature axonal and dendritic development (Duan et al., Reference Duan, Chang, Ge, Faulkner, Kim and Kitabatake2007; Eisch et al., Reference Eisch, Cameron, Encinas, Meltzer, Ming and Overstreet-Wadiche2008). Epigenetic mechanisms can also impact neuroplasticity. Epigenetics refers to heritable changes in gene expression that do not involve changes in actual DNA sequences. Three major mechanisms of epigenetic changes include DNA methylation, histone modification (such as acetylation), and noncoding RNAs (Hsieh & Eisch, Reference Hsieh and Eisch2010). Noncoding RNAs have been shown to regulate the proliferation of neural stem cells, either stimulating division of neural progenitor cells or promoting the apoptosis of cells (Iyengar et al., Reference Iyengar, Choudhary, Sarangdhar, Venkatesh, Gadgil and Pillai2014). Deficiency in growth arrest in DNA-damage-inducible beta (Gadd45b), a gene that promotes DNA demethylation, has been associated with deficits in neurogenesis and dendritic growth of neurons (Ma et al., Reference Ma, Jang, Guo, Kitabatake, Chang and Pow-Anpongkul2009). Late-phase LTP is dependent on gene transcription, which in turn depends on epigenetic processes. Thus, deletion of the gene coding for cAMP response element binding protein (CREB), which activates transcription through histone acetylation, results in impairments of late-phase LTP in animal models (Alarcon et al., Reference Alarcon, Malleret, Touzani, Vronskaya, Ishii and Kandel2004). In a related fashion, histone deacetylase inhibitors can enhance the induction of LTP by promoting gene transcription (Levenson & Sweatt, Reference Levenson and Sweatt2005).
LTP and LTD can also be impaired by various genetic mutations and deletions. For example, deletions in the genes coding for GluN2A subunit of the NMDA receptor (Kannangara et al., Reference Kannangara, Eadie, Bostrom, Morch, Brocardo and Christie2014) and subunits of calcium/calmodulin-dependent protein kinase II (Malenka & Nicoll, Reference Malenka and Nicoll1999) result in impairments in LTP. Numerous other genes that have been linked to LTP and LTD, including CREB1 (Bourtchuladze et al., Reference Bourtchuladze, Frenguelli, Blendy, Cioffi, Schutz and Silva1994), mammalian target of rapamycin (mTor; Hoeffer & Klann, Reference Hoeffer and Klann2010), and glycogen synthase kinase-3 beta (GSK-3B; Bradley et al., Reference Bradley, Peineau, Taghibiglou, Nicolas, Whitcomb and Bortolotto2012),
The Val66Met polymorphism in the BDNF gene results in the substitution of the amino acid valine with methionine, and is associated with changes in cortical morphology and hippocampal activity. The methionine allele has been associated with volumetric reductions in the hippocampus and prefrontal cortex (Pezawas et al., Reference Pezawas, Verchinski, Mattay, Callicott, Kolachana and Straub2004; Szeszko et al., Reference Szeszko, Lipsky, Mentschel, Robinson, Gunduz-Bruce and Sevy2005), and poorer performance in episodic memory tasks in normal individuals (Egan et al., Reference Egan, Kojima, Callicott, Goldberg, Kolachana and Bertolino2003). This polymorphism may affect plasticity by impairing NMDA-receptor mediated LTP (Ninan et al., Reference Ninan, Bath, Dagar, Perez-Castro, Plummer and Lee2010).
Environmental factors can substantially impact neurogenesis. Maternal infectious exposure in rats is associated with decreased neurogenesis in the dentate gyrus of the hippocampus (Cui, Ashdown, Luheshi, & Boksa, Reference Cui, Ashdown, Luheshi and Boksa2009) and diminished cognitive performance in offspring after birth (Jiang et al., Reference Jiang, Zhu, Zhao, Shen, Yu and Xu2013). This correlation may be mediated by immune system activation (De Miranda et al., Reference De Miranda, Yaddanapudi, Hornig, Villar, Serge and Lipkin2010). Prenatal exposure to substances of abuse, such as alcohol, can also lead to dysmorphic brain development (Gil-Mohapel, Boehme, Kainer, & Christie, Reference Gil-Mohapel, Boehme, Kainer and Christie2010). In addition, rodent models of chronic stress demonstrate decreases in hippocampal progenitor cells (Hsieh & Eisch, Reference Hsieh and Eisch2010; Pham, Nacher, Hof, & McEwen, Reference Pham, Nacher, Hof and McEwen2003).
While many variables have been observed to inhibit adult neurogenesis, other factors may enhance it. Deep brain stimulation, antidepressants, and exercise have been shown to increase adult neurogenesis in rodent models (Eisch et al., Reference Eisch, Cameron, Encinas, Meltzer, Ming and Overstreet-Wadiche2008). While promising, most research on this topic has been conducted on animal models. New techniques are being developed for in vivo visualization of neurogenesis in humans, such as the use of metabolic biomarkers to identify neural stem cells using proton magnetic resonance spectroscopy (Manganas et al., Reference Manganas, Zhang, Li, Hazel, Smith and Wagshul2007). Further technical advances may allow for direct study of neurogenesis in human neuropsychiatric illness.
Critical periods and timing of onset and closure of neuroplasticity
Environment can shape brain function substantively across the life span. Plasticity is at its greatest during key epochs early in development (critical periods), and this presents developmental psychopathologists with new avenues for understanding vulnerability of the brain and intervening in a timely manner (Cicchetti & Toth, Reference Cicchetti and Toth2009). Studies of critical periods in the visual cortex have shown that among other processes, maturation of specific GABA circuits may determine the onset of certain critical periods. Timing and duration of critical periods may be modifiable by pharmacological manipulation of these and similar circuits (Takesian & Hensch, Reference Takesian and Hensch2013). The onset of critical periods may be triggered by factors such as polysialic acid and neural cell adhesion molecule, which limit function of parvalbumin containing GABA circuits. Neural networks refined by experience are then actively stabilized by extracellular milieu factors, such as perineural nets, which serve as “brakes” for pruning processes (Wang & Fawcett, Reference Wang and Fawcett2012). An understanding of such factors is likely to shed light on disorders of neuroplasticity such as schizophrenia, and motivate potentially novel treatment targets (Bitanihirwe & Woo, Reference Bitanihirwe and Woo2014).
Effects of age: Plasticity across the life span
The brain maintains some plasticity throughout life, but the capacity to change, which is at its peak early in life, gradually declines with age after young adulthood. The degree, slope, and timing of such decline, however, varies between individuals, is determined by both genetic and environmental factors, and may underlie risk for neuropsychiatric disorders (Oberman & Pascual-Leone, Reference Oberman and Pascual-Leone2013).
Metaplasticity
The concept of metaplasticity refers to the plasticity of synaptic plasticity; that is, the ability of a synapse to engage in LTP or LTD can itself be modulated in a dynamic fashion (Abraham & Bear, Reference Abraham and Bear1996). Metaplasticity involves a priming stimulus that alters the subsequent response of a neuron to a plasticity-inducing stimulus. An important feature of metaplasticity is a time gap between the priming signal that stimulates metaplastic mechanisms and subsequent events that induce synaptic plasticity. Several mechanisms may enable metaplasticity. NMDA receptor activation, which induces LTP, also inhibits subsequent LTP for some time afterward (Abraham, Reference Abraham2008). Another mechanism may be metabotropic glutamate receptor activation, which enhances the induction of LTP in the hippocampus (Cohen, Coussens, Raymond, & Abraham, Reference Cohen, Coussens, Raymond and Abraham1999). Finally, mechanisms of nonsynaptic plasticity (i.e., intrinsic plasticity) may also be categorized as a type of metaplasticity (Abraham, Reference Abraham2008). One possible function of metaplasticity may be to protect against excitotoxic damage to neurons that could occur through unopposed LTP (Abraham, Reference Abraham2008).
Summary
The brain maintains plasticity throughout life, though in varying degrees at the different epochs of age. This remarkable ability of the brain is orchestrated by the inherent properties of neurons, synapses, and glia, and by neurotransmitter systems such as glutamate, GABA, and neurotrophic factors. As a result, cortical reorganization occurs in response to learning and to injury throughout life. The extent to which the brain can dynamically change with learning and exogenous exposures is determined by genetic, epigenetic, and environmental factors; such plastic change could be adaptive or represent maladaptive cascades secondary to genetic and environmental inputs, as we will see in the next section.
Plasticity Alterations in Schizophrenia
Schizophrenia is a common, chronic complex illness typically beginning in adolescence and characterized by positive symptoms (hallucinations, delusions, and disorganized thinking), negative symptoms (social withdrawal, affect flattening, and motivational deficits), and impaired cognition across several domains (attention, executive function, memory, and social cognition; Tandon, Keshavan, & Nasrallah, Reference Tandon, Keshavan and Nasrallah2008). It is widely held that schizophrenia is a developmental brain disorder, involving several processes affecting brain plasticity: early (neurogenesis, neural migration, and synaptogenesis; Murray, Lewis, Owen, & Foerster, Reference Murray, Lewis, Owen and Foerster1988; Weinberger, Reference Weinberger1987) and late in brain development (synaptic pruning, and myelination; Feinberg, Reference Feinberg1982; Keshavan, Anderson, & Pettegrew, Reference Keshavan, Anderson and Pettegrew1994; Murray et al., Reference Murray, Lewis, Owen and Foerster1988; Weinberger, Reference Weinberger1987). We herein review extant literature on alterations in schizophrenia that bear upon these neuroplasticity processes. There is evidence for diminished plasticity as well as aberrant excessive plasticity, as our review will show.
Neurons and synapses
Schizophrenia has been associated with a number of neuropathological abnormalities, which may also reflect deficiencies in plasticity. While neuroimaging studies demonstrate subtle reductions in gray matter volume in schizophrenia, postmortem studies indicate that this reduction is due to loss of cortical neuropil and dendritic arborization, rather than loss of neurons (Selemon, Mrzljak, Kleinman, Herman, & Goldman-Rakic, Reference Selemon, Mrzljak, Kleinman, Herman and Goldman-Rakic2003). The most consistent neuropathological findings in schizophrenia are reduced density of dendritic spines (Glantz & Lewis, Reference Glantz and Lewis2000; Harrison, Reference Harrison1999) and smaller cell bodies of neurons in the dorsolateral prefrontal cortex (Pierri, Volk, Auh, Sampson, & Lewis, Reference Pierri, Volk, Auh, Sampson and Lewis2001) and the hippocampus (Bennett, Reference Bennett2011). Adolescence, when schizophrenia typically begins, is a period during which the synaptic density is normally pruned by 50% (Anderson, Classey, Conde, Lund, & Lewis, Reference Anderson, Classey, Conde, Lund and Lewis1995; Woo, Reference Woo2013). Consequently, it has been suggested that dysfunctional or excessive synaptic pruning in the prefrontal cortex during adolescence may serve to diminish plasticity in schizophrenia (Keshavan et al., Reference Keshavan, Anderson and Pettegrew1994).
Altered neurotransmission
NMDA receptors and glutamatergic pathways play a crucial role in modulating synaptic plasticity (Butefisch et al., Reference Butefisch, Davis, Wise, Sawaki, Kopylev and Classen2000); they have also been implicated in schizophrenia (Moghaddam & Javitt, Reference Moghaddam and Javitt2012; Woo, Reference Woo2013) based on studies of NMDA antagonists causing psychotic and cognitive symptoms and electrophysiological changes in healthy individuals (Javitt, Steinschneider, Schroeder, & Arezzo, Reference Javitt, Steinschneider, Schroeder and Arezzo1996), and diminished cortical NMDA receptor subunit expression in individuals with schizophrenia (Harrison, Law, & Eastwood, Reference Harrison, Law and Eastwood2003). NMDA receptor hypofunction may cause glutamatergic excess and damage to pyramidal neurons, which may manifest as loss of dendritic arborization (Woo, Reference Woo2013), leading to diminished neuroplasticity.
Impairments in GABAergic systems may also disrupt plasticity in schizophrenia. Inhibitory parvalbumin-containing neurons promote the normal maturation of neuronal circuits, and are abnormal in schizophrenia (Woo, Reference Woo2013). In postmortem brains of schizophrenia patients, parvalbumin-containing neurons demonstrate diminished expression of glutamic acid decarboxylase 67 (GAD67), an enzyme that helps synthesize the inhibitory neurotransmitter GABA (Akbarian et al., Reference Akbarian, Kim, Potkin, Hagman, Tafazzoli and Bunney1995). Thus, in patients with schizophrenia, these neurons may fail to regulate synaptic pruning. GABA activity may be decreased in certain brain regions in schizophrenia (Barr et al., Reference Barr, Farzan, Rajji, Voineskos, Blumberger and Arenovich2013), which may lead to reduced cortical plasticity (Butefisch et al., Reference Butefisch, Davis, Wise, Sawaki, Kopylev and Classen2000; Voineskos, Rogasch, Rajji, Fitzgerald, & Daskalakis, Reference Voineskos, Rogasch, Rajji, Fitzgerald and Daskalakis2013) and abnormal pruning. In addition, maturation of the extracellular matrix, comprising perineuronal nets, may be critical for termination of synaptic pruning processes (Woo, Reference Woo2013); failure of such maturation may lead to “runaway” pruning.
Glial alterations
Alterations in microglia, a type of neuronal support cell, may also contribute to impaired plasticity in schizophrenia. Both imaging and postmortem studies have observed increased activation of microglia in patients with schizophrenia. This has been noted in both the frontal cortex and the hippocampus (Doorduin et al., Reference Doorduin, de Vries, Willemsen, de Groot, Dierckx and Klein2009). In an analysis of publicly available gene pathways related to glial cell function from the Psychiatric Genomics Consortium data, the glia–oligodendrocyte pathway was specifically associated with schizophrenia, indicating how oligodendrocyte dysfunction may contribute to the myelination abnormalities seen in schizophrenia (Duncan et al., Reference Duncan, Holmans, Lee, O'Dushlaine, Kirby and Smoller2014).
Alterations in neurotrophins
Deficits in neurotrophins, particularly BDNF, may underlie diminished plasticity in schizophrenia. Levels of BDNF are lower in schizophrenia than in healthy controls and are associated with severity of both positive (Pillai et al., Reference Pillai, Kale, Joshi, Naphade, Raju and Nasrallah2010) and negative symptoms (Chen et al., Reference Chen, Lee, Chang, Chen, Chu and Wang2014). As discussed earlier, the Val66Met polymorphism may impair the cellular transport of BDNF. Data on the association of this polymorphism with schizophrenia is inconsistent. One meta-analysis found that homozygosity for the infrequent methionine/methionine genotype was associated with elevated risk of schizophrenia (Gratacos et al., Reference Gratacos, Gonzalez, Mercader, de Cid, Urretavizcaya and Estivill2007), though other meta-analyses have not confirmed this finding (Kanazawa, Glatt, Kia-Keating, Yoneda, & Tsuang, Reference Kanazawa, Glatt, Kia-Keating, Yoneda and Tsuang2007; Naoe et al., Reference Naoe, Shinkai, Hori, Fukunaka, Utsunomiya and Sakata2007).
BDNF appears to have an intricate relationship with the dopamine neurotransmitter system. While BDNF mediates expression of D1 and D5 dopamine receptors, removal of dopaminergic neurons in the midbrain is associated with diminished levels of BDNF, suggesting that these neurons influence BDNF gene expression (Favalli, Belmonte-de-Abreu, Wong, & Daskalakis, Reference Favalli, Li, Belmonte-de-Abreu, Wong and Daskalakis2012). BDNF levels may rise with antipsychotic treatment, though again, evidence is inconsistent (Favalli et al., Reference Favalli, Li, Belmonte-de-Abreu, Wong and Daskalakis2012; Grillo et al., Reference Grillo, Ottoni, Leke, Souza, Portela and Lara2007).
Abnormal sleep spindles and EEG findings
Patients with schizophrenia demonstrate significant reductions in density, number, and coherence of sleep spindles. Synchronous oscillations of neural circuits during spindle sleep have been thought to contribute to learning-related synaptic plasticity. Motor procedural learning during sleep, normally seen in healthy individuals, is impaired in schizophrenia, and this deficit is related to spindle reductions. (Wamsley et al., Reference Wamsley, Tucker, Shinn, Ono, McKinley and Ely2012). Spindle reductions appear to be related to cognitive impairments in early course patients with schizophrenia (Keshavan, Montrose, Miewald, & Jindal, Reference Keshavan, Montrose, Miewald and Jindal2011).
Some EEG abnormalities seen in schizophrenia may reflect impaired plasticity. For example, prepulse inhibition of the startle response refers to a decrease in the amplitude of the startle response that occurs when the startling stimulus is preceded by a weak stimulus. In schizophrenia, the startle response does not decrease as much as it does in healthy controls (Braff, Geyer, & Swerdlow, Reference Braff, Geyer and Swerdlow2001), implying failure of habituation to a stimulus. This diminished habituation may reflect abnormal plasticity, in that the brain is unable to efficiently adapt to environmental change. Mismatch negativity, which represents an evoked response to an unexpected deviant stimulus, is also impaired in schizophrenia, and has been thought to reflect abnormal NMDA mediated short-term plasticity.
Diminished LTP and LTD-like network plasticity
As reviewed in the earlier section, functional MRI studies have demonstrated evidence of cortical plasticity in humans. This cortical remapping is observed in both healthy individuals following motor learning tasks and subjects with brain injury. It is also believed to underlie important perceptual and motor learning abilities (Reed et al., Reference Reed, Riley, Carraway, Carrasco, Perez and Jakkamsetti2011). The cellular substrate of such cortical map plasticity is hypothesized to be related to the better demonstrated synaptic plasticity (Buonomano & Merzenich, Reference Buonomano and Merzenich1998). Reduced neuroplasticity in schizophrenia could lead to deficit states such as cognitive deficits, negative symptoms, and functional disability (Fett et al., Reference Fett, Viechtbauer, Dominguez, Penn, van Os and Krabbendam2011; Green et al., Reference Green, Nuechterlein, Gold, Barch, Cohen and Essock2004; Sergi et al., Reference Sergi, Rassovsky, Widmark, Reist, Erhart and Braff2007). Evidence to support reduced cortical plasticity in schizophrenia comes from novel neuroimaging experiments that incorporate brain stimulation and EEGs.
Transcranial magnetic stimulation (TMS) has been used to study in vivo cortical plasticity in schizophrenia. This method uses focal magnetic fields to penetrate the cranium. The resultant electric currents then depolarize the underlying cortex, thus inducing action potentials in targeted brain regions (Kobayashi & Pascual-Leone, Reference Kobayashi and Pascual-Leone2003). The output of cortical activation (in this case motor cortex) is measured using electromyographic recordings of hand muscle contractions. The most common method has been to compare motor evoked potentials (MEP) and motor thresholds before and after repetitive brain stimulation, with repetitive TMS (rTMS) or transcranial direct current stimulation (tDCS), which uses direct currents to shift the resting membrane potentials of underlying neurons (Nitsche & Paulus, Reference Nitsche and Paulus2000). These techniques use synaptic plasticity-inducing protocols that result in cortical excitability changes mirroring LTP (high-frequency rTMS and anodal tDCS) or LTD (low-frequency rTMS and cathodal tDCS).
Compared to healthy controls, reduced LTD-like plasticity has been reported in schizophrenia patients by demonstrating lack of expected changes in MEP (reduction in amplitude) and motor thresholds (increase) as induced by low-frequency rTMS, delivered to the premotor (Oxley et al., Reference Oxley, Fitzgerald, Brown, de Castella, Daskalakis and Kulkarni2004) and motor (Fitzgerald et al., Reference Fitzgerald, Brown, Marston, Oxley, De Castella and Daskalakis2004) cortices, as well as by cathodal tDCS delivered to the motor cortex (Hasan, Nitsche, et al., Reference Hasan, Nitsche, Herrmann, Schneider-Axmann, Marshall and Gruber2012). It is interesting that these deficits were also demonstrated in recordings from the nonstimulated hemisphere (Hasan, Aborowa, et al., Reference Hasan, Aborowa, Nitsche, Marshall, Schmitt and Gruber2012), suggesting an association between LTD-like cortical plasticity and interhemispheric connectivity.
Possible impairments in LTP-like plasticity have also been demonstrated using similar study designs. Lesser enhancement of MEP was observed after anodal tDCS to the contralateral motor cortex in chronic schizophrenia patients relative to recent-onset patients and healthy controls (Hasan et al., Reference Hasan, Nitsche, Rein, Schneider-Axmann, Guse and Gruber2011). Frantseva et al. (Reference Frantseva, Fitzgerald, Chen, Moller, Daigle and Daskalakis2008) used a different strategy to measure LTP-like plasticity by pairing (within 25 ms) median nerve electric stimulation with TMS over the contralateral motor cortex, in what is referred to as paired associative stimulation. Schizophrenia patients showed lesser facilitation of MEPs, when compared to healthy individuals. It is interesting that these patients also demonstrated motor learning deficits, and there was a significant association between the measure of LTP and motor skill learning. Use-dependent plasticity is another TMS measure that may reflect LTP-like plasticity (Classen, Liepert, Wise, Hallett, & Cohen, Reference Classen, Liepert, Wise, Hallett and Cohen1998). Here, the spontaneous direction of TMS-induced thumb movements is first measured. Subjects are then trained with 30 min of motoric practice of thumb movements in a direction that is opposite (by 180 degrees) to the actual thumb movements. Postpractice thumb movement direction elicited by TMS is then evaluated. Using this experiment, Daskalakis, Christensen, Fitzgerald, and Chen (Reference Daskalakis, Christensen, Fitzgerald and Chen2008) found that schizophrenia patients had significantly attenuated motor reorganization compared to healthy subjects.
Stimulus-specific plasticity paradigms using event-related potentials have also been used to quantify occipital (visual) and temporal (auditory) lobe LTP-like plasticity (Clapp, Kirk, et al., Reference Clapp, Kirk, Hamm, Shepherd and Teyler2005; Clapp, Zaehle, et al., Reference Clapp, Zaehle, Lutz, Marcar, Kirk and Hamm2005). Here, repetitive high-frequency visual or auditory stimuli are used to produce a lasting facilitation of visual or auditory evoked potentials, respectively. Using this paradigm, researchers have demonstrated lesser facilitation of visual (Cavus et al., Reference Cavus, Reinhart, Roach, Gueorguieva, Teyler and Clapp2012) and auditory (Mears & Spencer, Reference Mears and Spencer2012) evoked potentials in schizophrenia patients as compared to healthy controls.
Overall, these findings not only provide evidence for deficient cortical plasticity that represent both LTD and LTP-like synaptic plasticity but also link these deficits to impairments in cognitive functions like learning and memory (Frantseva et al., Reference Frantseva, Fitzgerald, Chen, Moller, Daigle and Daskalakis2008; Wamsley et al., Reference Wamsley, Tucker, Shinn, Ono, McKinley and Ely2012).
Genes, environment, and impaired plasticity in schizophrenia
Schizophrenia is highly heritable. In recent years, several genetic loci with small to moderate effects have been identified in genomewide association studies. It is interesting that these genes not only regulate glutamatergic, GABAergic, and dopaminergic transmission but also regulate several aspects of brain development and plasticity discussed above (Balu & Coyle, Reference Balu and Coyle2011). Alterations in the DISC1 gene, expressed during both prenatal and adult hippocampal neurogenesis (Jun, Hussaini, Rigby, & Jang, Reference Jun, Mohammed Qasim Hussaini, Rigby and Jang2012) have demonstrated signs of maladaptive plasticity (mistargeted formation of synapses and reduced dendritic arborization) in mice (Kvajo et al., Reference Kvajo, McKellar, Drew, Lepagnol-Bestel, Xiao and Levy2011; Pletnikov et al., Reference Pletnikov, Ayhan, Nikolskaia, Xu, Ovanesov and Huang2008). Time-specific transient alterations (e.g., in utero) of DISC1 have shown to adversely affect postnatal maturation of prefrontal dopaminergic and GABAergic neurotransmission (Niwa et al., Reference Niwa, Kamiya, Murai, Kubo, Gruber and Tomita2010). The neuregulin 1 gene (NRG1), which codes for the protein neuregulin 1, is involved in adult neurogenesis. NRG1 has been shown to stimulate proliferation of hippocampus-derived neural progenitor cells (Lai & Feng, Reference Lai and Feng2004), and partial deletions of this gene are associated with stress sensitivity in animal models (Chohan et al., Reference Chohan, Boucher, Spencer, Kassem, Hamdi and Karl2014). Thus, genetic alterations in the capacity for neurogenesis may weaken the brain's response to environmental stress, elevating the risk for development of neuropsychiatric disorders.
One novel line of work has used human induced pluripotent stem cells to examine alterations in neurogenesis in schizophrenia. In this method, fibroblasts are obtained from individuals and reprogrammed into pluripotent stem cells. In one such study in patients with schizophrenia, these neurons displayed a significant decrease in the number of neurites and neuronal connectivity (Brennand et al., Reference Brennand, Simone, Jou, Gelboin-Burkhart, Tran and Sangar2011). Abnormalities in gene expression were also observed, such as decreased expression of the protein PSD95 and increased expression of NRG1. Of the several hundred genes that demonstrated abnormal expression, 13% were reported to be abnormal in schizophrenia in previous publications (Brennand et al., Reference Brennand, Simone, Jou, Gelboin-Burkhart, Tran and Sangar2011).
Environmental factors may mediate the dendritic spine reductions observed in schizophrenia. Chronic stress and prenatal stress have been correlated with reduced dendritic arborization in animal models (Markham, Mullins, & Koenig, Reference Markham, Mullins and Koenig2013), while environmental enrichment and learning are associated with increased dendritic arborization (O'Malley, O'Connell, Murphy, & Regan, Reference O'Malley, O'Connell, Murphy and Regan2000). In summary, plasticity in schizophrenia may be abnormal due to genetically mediated changes in NMDA receptor function, GABA-mediated inhibition, and neurogenesis. These abnormalities eventually lead to observable neuropathological abnormalities in dendritic spine density and complexity. Epigenetic factors may mediate the impact of environmental factors on plasticity processes via noncoding RNAs (Spadaro & Bredy, Reference Spadaro and Bredy2012).
Aberrant excessive neuroplasticity in schizophrenia?
In contrast to diminished plasticity, aberrant excessive synaptic plasticity in neural networks may underlie positive symptoms of schizophrenia; this may result from dysregulated metaplasticity secondary to either genetically controlled reduced synaptic plasticity in key cortical regions or environmental effects like stress or substance abuse. For instance, Hoffman has suggested that social withdrawal or “deafferentation” may trigger the initial active phase of schizophrenia (Hoffman, Reference Hoffman2007) by plastic reorganization by the “social brain” to generate spurious meaning from social cues that may manifest as hallucinations or delusions (Hoffman, Reference Hoffman2008). This may reflect metaplastic effects (Abraham, Reference Abraham2008) on social brain regions. Animal experiments suggest that social isolation enhances the surface trafficking of NMDA receptors in dendritic spines of principal neurons in the amygdala, thus leading to aberrant plasticity and emotion dysregulation (Gan, Bowline, Lourenco, & Pickel, Reference Gan, Bowline, Lourenco and Pickel2014). These findings are also in sync with the observation that sensory deafferentation induces hyperplastic brain changes that may be mediated either by removal of GABA-related cortical inhibition or by LTP-like mechanisms (Ziemann, Hallett, & Cohen, Reference Ziemann, Hallett and Cohen1998).
Support for aberrant excessive plasticity also comes from neuroimaging studies examining the dysconnection hypothesis of schizophrenia (Stephan, Friston, & Frith, Reference Stephan, Friston and Frith2009). Diffusion tensor imaging has revealed greater white matter connectivity in schizophrenia patients with auditory hallucinations, in contrast to those without in the arcuate fasciculus, which connects the primary auditory cortex with language areas, and the cingulate bundle, a part of the limbic cortex (Hubl et al., Reference Hubl, Koenig, Strik, Federspiel, Kreis and Boesch2004). This aberrant connectivity could underlie the abnormal coactivation of regions that normally process external auditory stimuli and language-related areas (Dierks et al., Reference Dierks, Linden, Jandl, Formisano, Goebel and Lanfermann1999). Moreover, patients with both auditory and visual hallucinations show higher white matter connectivity in the pathways connecting the visual areas to the hippocampal formation (Amad et al., Reference Amad, Cachia, Gorwood, Pins, Delmaire and Rolland2014) and the amygdala (Ford et al., Reference Ford, Palzes, Roach, Potkin, van Erp and Turner2014), when compared to patients with only auditory hallucinations. Similarly, patients with auditory hallucinations show increased resting-state functional connectivity between the hippocampal formation and the language regions (Sommer, Clos, Meijering, Diederen, & Eickhoff, Reference Sommer, Clos, Meijering, Diederen and Eickhoff2012). These findings partially support earlier observations of heightened regional hippocampal blood flow in schizophrenia patients at rest and during a cognitive task (Medoff, Holcomb, Lahti, & Tamminga, Reference Medoff, Holcomb, Lahti and Tamminga2001) and emerging evidence on correlations between hippocampal volumes and psychotic symptoms (Mathew et al., Reference Mathew, Gardin, Tandon, Eack, Francis and Seidman2014). In another study that combined resting-state and task (working memory) based functional imaging, patients with schizophrenia and their relatives demonstrated hyperactivation (reduced task-related suppression) and hyperconnectivity of the default mode network (Whitfield-Gabrieli et al., Reference Whitfield-Gabrieli, Thermenos, Milanovic, Tsuang, Faraone and McCarley2009) when compared to healthy subjects. These abnormalities were associated with severity of psychopathology and cognitive deficits (Whitfield-Gabrieli et al., Reference Whitfield-Gabrieli, Thermenos, Milanovic, Tsuang, Faraone and McCarley2009). Finally, structural MRI studies have demonstrated significantly increased cortical thickness in regions related to self-monitoring (the left insular cortex, cingulate gyrus, and dorsal middle frontal gyrus and hippocampal formation) in schizophrenia patients with auditory hallucinations than those without (Amad et al., Reference Amad, Cachia, Gorwood, Pins, Delmaire and Rolland2014; van Swam et al., Reference van Swam, Federspiel, Hubl, Wiest, Boesch and Vermathen2012). It is interesting that auditory hallucinations have been linked to a failure to activate areas concerned with the monitoring of inner speech (McGuire et al., Reference McGuire, Silbersweig, Wright, Murray, David and Frackowiak1995); impaired corollary discharges, which are neural signals that coincide with self-generated thoughts/movements (Crapse & Sommer, Reference Crapse and Sommer2008), may underlie increased cortical activity to self-induced sensory stimuli observed in patients with schizophrenia (Whitford et al., Reference Whitford, Mathalon, Shenton, Roach, Bammer and Adcock2011). It has been speculated that the mirror neuron system plays a role in the generation of these corollary discharges (Prather, Peters, Mowicki, & Mooney, Reference Prather, Peters, Nowicki and Mooney2008; Tchernichovski & Wallman, Reference Tchernichovski and Wallman2008). Mirror neuron system activity is reduced in schizophrenia (Kato et al., Reference Kato, Muramatsu, Kato, Shibukawa, Shintani and Mimura2011; Mehta, Agarwal, et al., Reference Mehta, Agarwal, Kalmady, Shivakumar, Kumar and Venkatasubramanian2013). These deficits were also found to be associated with social cognitive deficits in these patients (Mehta, Basavaraju, Thirthalli, & Gangadhar, Reference Mehta, Basavaraju, Thirthalli and Gangadhar2012; Mehta, Thirthalli, Bassavaraju, Gangadhar, & Pascual-Leone, Reference Mehta, Basavaraju, Thirthalli and Gangadhar2013).
The 22q11 microdeletion syndrome is the strongest known link between any genetic anomaly and schizophrenia, with as many as 30% developing symptoms of schizophrenia (Pulver et al., Reference Pulver, Nestadt, Goldberg, Shprintzen, Lamacz and Wolyniec1994). Mouse models of the 22q11 microdeletion syndrome show a dramatic enhancement in short- and long-term potentiation of synaptic transmission in an age-dependent manner in the hippocampus of these mice, as compared to the wild-type mice (Earls et al., Reference Earls, Bayazitov, Fricke, Berry, Illingworth and Mittleman2010). The 22q11 microdeletion may lead to a reduction in the Dgcr8 gene, resulting in an elevated Serca2 expression causing abnormally excessive synaptic plasticity (Earls & Zakharenko, Reference Earls and Zakharenko2013). Another mechanism through which 22q11 microdeletion syndrome manifests is haploinsufficiency of the transcription factor TBX1. This transcription factor interacts with several signaling pathways, including β-catenin, a protein that functions as the “molecular glue” to keep synapses together (Papangeli & Scambler, Reference Papangeli and Scambler2013). Recent evidence from mice experiments has demonstrated that excessive hippocampal beta-catenin can potentially lead to “sticky synapses” that have impaired LTD-like plasticity, which result in impaired cognitive flexibility (Mills et al., Reference Mills, Bartlett, Dissing-Olesen, Wisniewska, Kuznicki and Macvicar2014). This process may yield itself as a mechanistic basis to understand the inflexible nature of persistent delusions in schizophrenia.
As reviewed earlier, mutations in the DISC1 gene are considered risk factors for schizophrenia (Harrison & Weinberger, Reference Harrison and Weinberger2005). DISC1 knockdown models have demonstrated an accelerated hippocampal neurogenesis, as well as increased dendritic development and synapse formation. These aberrant morphological changes result in an accelerated formation of functional GABAergic and glutamatergic synaptic inputs to new neurons, as well as enhanced excitability of the hippocampal neurons (Duan et al., Reference Duan, Chang, Ge, Faulkner, Kim and Kitabatake2007). DISC1 thus appears to be a critical regulator of the aberrant excessive synaptic plasticity observed in schizophrenia. Yet another animal model of schizophrenia that employs phospholipase C-β1 knockout mice has also demonstrated significantly enhanced adult hippocampal neurogenesis in these mice when compared with the wild-type littermates (Manning, Ransome, Burrows, & Hannan, Reference Manning, Ransome, Burrows and Hannan2012).
Together, the above synthesis of evidence for aberrant excessive synaptic plasticity in limbic regions against a background of reduced cortical plasticity provides a framework to understand different symptom dimensions of schizophrenia within the broad purview of the “dysplastic” model.
Summary
Several lines of evidence point to diminished neuronal plasticity in widespread brain regions in schizophrenia, including reductions in dendritic and glial density; altered glutamatergic, GABAergic, and neurotrophic function; and in vivo evidence of diminished LTP and LTD-like plasticity. While these changes may account for the core deficit symptoms of schizophrenia, positive symptoms might result from excessive neuroplasticity causing aberrant reorganization in limbic circuits. Genetic, epigenetic, and environmental factors, as discussed below, may influence the nature, extent, timing, and persistence of such abnormalities.
Aberrant Plasticity and the Schizophrenia Risk State
Schizophrenia has been linked to a plethora of genetic as well as socioenvironmental risk factors (Morgan, McKenzie, & Fearon, Reference Morgan, McKenzie and Fearon2008; Shah, Mizrahi, & McKenzie, Reference Shah, Mizrahi and McKenzie2011; Sullivan, Reference Sullivan2005), which may impact (either directly or indirectly) the kinds of neuroplastic processes described in previous sections. The extant neurodevelopmental hypotheses of schizophrenia (Fatemi & Folsom, Reference Fatemi and Folsom2009; Keshavan, Reference Keshavan1999; McGrath, Féron, Burne, Mackay-Sim, & Eyles, Reference McGrath, Féron, Burne, Mackay-Sim and Eyles2003; Murray, Reference Murray1994) suggest that developmental brain changes may occur during the prenatal or in postnatal life extending into adolescence or early adulthood. Such alterations could profoundly alter early brain developmental processes such as neuronal proliferation, migration, apoptosis, and synaptogenesis, and/or later processes of synaptic pruning and myelination. These processes are further impacted by hormonal changes, and exogenous insults such as trauma, neglect, and substance abuse during childhood or adolescence (Keshavan, Reference Keshavan1999; Paus, Keshavan, & Giedd, Reference Paus, Keshavan and Giedd2008; Piper et al., Reference Piper, Beneyto, Burne, Eyles, Lewis and McGrath2012). As the combination of “hits” individuals encounter increases, their brains are vulnerable to becoming prone to more distressing symptoms and worsening functional impairment (Owen, Donovan, Thapar, & Craddock, Reference Owen, Donovan, Thapar and Craddock2011). The role of certain chronic risk factors (such as negative life events, daily hassles, or substance misuse) appears to be additive and cumulative (Collip, Myin-Germeys, & van Os, Reference Collip, Myin-Germeys and van Os2008). We suggest that it is not only the cumulative adverse exposures per se but also repeated exposure that plays a role via altered brain plasticity in promoting liability to brain changes, symptoms, and impairment.
Trajectory of premorbid psychopathology in high-risk subjects may be linked to critical periods of vulnerability
If insults to core neurobiological processes contribute to the schizophrenia phenotype many years later, the neurodevelopmental hypothesis holds that such processes are both unfolding and especially susceptible to perturbation during critical periods. These periods may include specific windows of prenatal life, childbirth, childhood, and adolescence at which particular risk exposures have been identified. At the earliest stages of conception, for example, advanced paternal age, presumably through de novo mutations, appears to confer susceptibility (Malaspina, Reference Malaspina2001). During the prenatal period, maladaptive infectious exposure may influence maternal immune response and/or fetal physiology in an experience-expectant fashion to contribute to a modest but still significant risk for psychosis (Brown & Derkits, Reference Brown and Derkits2010). In childhood and adolescence, experience-dependent factors such as abuse, use of psychotropic substances and bullying also increase risk (Addington et al., Reference Addington, Stowkowy, Cadenhead, Cornblatt, McGlashan and Perkins2013; Shah et al., Reference Shah, Eack, Montrose, Tandon, Miewald and Prasad2012; van Dam et al., Reference van Dam, van der Ven, Velthorst, Selten, Morgan and de Haan2012).
The concept of combinations of exposures/insults at specific critical periods maps onto observations that the timing and course of development may differ across brain regions or circuits (Lewis & Akil, Reference Lewis and Akil1997). For example, synaptic density in the visual cortex reaches adult levels by preschool age (Toga, Thompson, & Sowell, Reference Toga, Thompson and Sowell2006), whereas higher order disruption in executive function has repeatedly been localized to the prefrontal cortex, an area that is among the last to complete maturation (Gogtay, Reference Gogtay2008). If endogenous or exogenous insults lead to disrupted neural processes that compromise neuroplasticity, then phenotypic manifestations may reflect the neural circuits maximally affected by failed, aberrant, or excessive plasticity.
The earliest detectable phases of psychotic illness (i.e. the component signs and symptoms with which it is consistently associated) tend to emerge after, rather than before, the accumulation of critical periods of exposure and brain maturation. This has allowed researchers to focus new attention on the trajectory of the premorbid and subthreshold stages in children and adolescents at high clinical and/or familial risk for schizophrenia. Individuals at familial high risk (FHR) have an 8%–12% chance of converting to psychosis over the life span; in contrast, in clinical studies, distressed and help-seeking clinical high-risk (CHR) subjects have a 15%–40% rate of conversion over a 2- to 3-year period (Fusar-Poli et al., Reference Fusar-Poli, Bechdolf, Taylor, Bonoldi, Carpenter and Yung2013; Tandon, Keshavan, & Nasrallah, Reference Tandon, Keshavan and Nasrallah2008). However, a broad spectrum of nonpsychotic psychopathology is substantively more common in the premorbid phase. In an ongoing prospective study of FHR relatives (Shah et al., unpublished data), we have observed that cognitive/learning disorders appear to emerge earliest, followed by anxiety and affective disorders, before social withdrawal and subthreshold psychotic-like symptoms and impaired cognition set in (Figure 4). The emergence of such a “classic” trajectory may reflect that the neural circuits underlying attention and sensorimotor function mature (i.e., show diminishing plasticity) earliest, followed by the maturation of limbic/striatal and eventually higher association brain regions such as the prefrontal cortex. However, as will be discussed further, the sequence, timing, and slope of such trajectory may vary greatly between individuals, in relation to genetic and environmental factors.

Figure 4. (Color online) Trajectory of phenotypic manifestations among individuals deemed to be at high risk for schizophrenia and the genetic and environmental predisposing factors.
Neuroplasticity may be altered in subjects at risk for psychosis
We herein review the relatively limited evidence suggesting that neurodevelopmental processes involved in schizophrenia etiopathogenesis reflect not only an altered trajectory of otherwise determined brain development but also disruptions to neuroplastic processes themselves.
Gray matter changes
While few neuropathological data exist, progressive gray matter loss seen in high-risk subjects indirectly suggests alterations in brain plasticity. CHR individuals who later developed psychosis had decreased gray matter volumes in some neuroanatomical structures compared with controls and nonconverters, as well as gray matter reductions over time (Borgwardt et al., Reference Borgwardt, McGuire, Aston, Berger, Dazzan and Gschwandtner2007; Pantelis et al., Reference Pantelis, Velakoulis, McGorry, Wood, Suckling and Phillips2003). However, in young high-risk siblings who did not develop serious psychopathology, gray matter deficits normalize by late adolescence, suggesting that normal plasticity is associated with resilience (Gogtay et al., Reference Gogtay, Greenstein, Lenane, Clasen, Sharp and Gochman2007; Mattai et al., Reference Mattai, Weisinger, Greenstein, Stidd, Clasen and Miller2011). Genetic and environmental factors may determine variation in trajectories of high-risk individuals (Peper, Brouwer, Boomsma, Kahn, & Hulshoff Pol, Reference Peper, Brouwer, Boomsma, Kahn and Hulshoff Pol2007). For example, more severe gray matter loss over time is seen in FHR subjects exposed to cannabis compared with those without such exposure (Welch et al., Reference Welch, Moorhead, McIntosh, Owens, Johnstone and Lawrie2013). Interaction between cortical thickness and the well-studied cathechol-O-methyltransferase (COMT) val158met polymorphism provides another example of differential susceptibility: while valine/valine homozygosity was related to steeper gray matter loss in adolescence in probands and their siblings, it attenuated cortical thinning in healthy controls (Raznahan et al., Reference Raznahan, Greenstein, Lee, Long, Clasen and Gochman2011).
Alterations in functional connectivity
FMRI studies in FHR subjects using nonlinear dynamic causal modelling have shown reduced thalamocortical connectivity (Dauvermann et al., Reference Dauvermann, Whalley, Romaniuk, Valton, Owens and Johnstone2013). In a recent fMRI study using an emotion recognition paradigm, Gee et al. (Reference Gee, Karlsgodt, van Erp, Bearden, Lieberman and Belger2012) demonstrated that relative to controls, CHR subjects showed increased amygdala and decreased ventral prefrontal cortex activation with age. This suggests that a failure of the prefrontal cortex to regulate amygdala reactivity emerges during adolescence and young adulthood.
Altered glutamatergic neurotransmission
Altered glutamatergic function implicated in impaired brain development (Keshavan, Reference Keshavan1999) may be related to aberrant neuroplasticity in schizophrenia. Neuronal “dysconnectivity,” in this model, may be mediated by abnormal NMDA receptor function. Evidence for this model stems from structural and functional neuroimaging, electroencephalography, neurophysiology, neuropharmacology, genetics, network modeling, neuropathology, and postmortem studies of patients with schizophrenia (Stephan et al., Reference Stephan, Friston and Frith2009). However, direct evidence of premorbid glutamatergic abnormalities is sparse. Of interest, recent magnetic resonance spectroscopy reports have found abnormal glutamine/glutamate levels in FHR and CHR subjects (de la Fuente-Sandoval et al., Reference de la Fuente-Sandoval, León-Ortiz, Favila, Stephano, Mamo and Ramírez-Bermúdez2011; Fusar-Poli et al., Reference Fusar-Poli, Stone, Broome, Valli, Mechelli and McLean2011; Stone et al., Reference Stone, Day, Tsagaraki, Valli, McLean and Lythgoe2009, Reference Stone, Howes, Egerton, Kambeitz, Allen and Lythgoe2010; Tandon et al., Reference Tandon, Bolo, Sanghavi, Mathew, Francis and Stanley2013).
Mismatch negativity, a promising biomarker in schizophrenia (Javitt et al., Reference Javitt, Steinschneider, Schroeder and Arezzo1996), is significantly reduced in CHR subjects compared to healthy control subjects and in those who convert to psychotic disorders (Perez et al., Reference Perez, Woods, Roach, Ford, McGlashan and Srihari2014). The mismatch negativity effect has been thought to reflect abnormal modulation of NMDA receptor-dependent plasticity (Baldeweg & Hirsch, Reference Baldeweg and Hirsch2014). Another physiological measure thought to reflect integrity of GABAergic circuits is that of gamma synchrony, known to be abnormal in schizophrenia. Recent evidence suggests abnormalities in gamma band responses to auditory stimuli in CHR subjects (Perez et al., Reference Perez, Woods, Roach, Ford, McGlashan and Srihari2014). These observations support the view that impaired NMDA/GABA mediated plasticity may underlie the risk for schizophrenia.
BDNF
As discussed earlier, neurotrophins such as BDNF appear to be altered in schizophrenia (Buckley, Pillai, & Howell, Reference Buckley, Pillai and Howell2011). In FHR groups, a verbal memory task undertaken during fMRI found decreased activation during encoding and retrieval in multiple corticolimbic regions for valine/valine BDNF homozygotes at the rs6265 polymorphism (Baig et al., Reference Baig, Whalley, Hall, McIntosh, Job and Cunningham-Owens2010). It is interesting that this risk allele does not suppress task-related frontal activation per se, because the same risk allele increased activation at the anterior cingulate cortex during a sentence completion task (Whalley et al., Reference Whalley, Baig, Hall, Job and McIntosh2010). Altogether, these variable findings suggest impaired neuroplasticity in frontal brain functions depending on region and/or task.
Increased stress reactivity and emergent symptoms may be related to aberrant plasticity
Adolescence denotes a period of increasing physiologic response to stress, which, if dysregulated, can alter the long-term set point of neurobiologic pathways (Walker, Sabuwalla, & Huot, Reference Walker, Sabuwalla and Huot2004). One mechanism invoked to explain the linkage between stress and psychotic symptoms is that of sensitization, the notion that repeated exposures to stress will produce successively larger physiologic responses over time, in some cases becoming aberrant, particularly in those at risk (Collip et al., Reference Collip, Myin-Germeys and van Os2008). First, recurrent administration and withdrawal of amphetamine in peripubertal mice leads to long-lasting alterations in neuroplasticity-related genes, which then may increase dopamine-dependent behaviors (Calabrese et al., Reference Calabrese, Richetto, Racagni, Feldon, Meyer and Riva2013). Support for this theory is found in experiments in which dopamine response following amphetamine challenge is associated with psychotic symptoms; in time, exposure to even attenuated stressors can lead to excessive dopamine release (Laruelle & Abi-Dargham, Reference Laruelle and Abi-Dargham1999; Lieberman, Sheitman, & Kinon, Reference Lieberman, Sheitman and Kinon1997). Second, dynamic changes in the hypothalamic–pituitary–adrenal (HPA) axis are believed to occur in response to internal and external stimuli and demands, whether adaptive or pathologic (Pariante, Reference Pariante2008). The number and significance of stressful life events increases as children enter adolescence (Gunnar & Quevedo, Reference Gunnar and Quevedo2007; Gunnar & Talge, Reference Gunnar, Talge, Schmidt and Segalowitz2011); with this comes HPA axis alterations, including higher basal cortisol levels and more robust acute responses to stress (Lupien, McEwen, Gunnar, & Heim, Reference Lupien, McEwen, Gunnar and Heim2009; Walker et al., Reference Walker, Trotman, Pearce, Addington, Cadenhead and Cornblatt2013). Pituitary volume is elevated in the early phases of the illness (first episode and high-risk subjects who later convert; Nordholm et al., Reference Nordholm, Krogh, Mondelli, Dazzan, Pariante and Nordentoft2013). Cortisol has also been repeatedly found to be elevated in psychosis (Borges, Gayer-Anderson, & Mondelli, Reference Borges, Gayer-Anderson and Mondelli2013) and in CHR subjects, particularly those who will later convert to psychosis (Sugranyes, Thompson, & Corcoran, Reference Sugranyes, Thompson and Corcoran2012; Walker et al., Reference Walker, Brennan, Esterberg, Brasfield, Pearce and Compton2010). Third, prevailing theories regard dopaminergic hyperactivity as a “final common pathway” by which attenuated and (later) full-blown symptoms emerge in psychosis. It has been postulated that dopaminergic dysregulation in psychosis is sensitized (influenced by stress) through the HPA axis, because mesolimbic dopamine activity is known to be associated with cortisol release, symptom appearance, and relapse. Positron emission tomography studies link psychosocial stress with dopamine release abnormalities in healthy individuals (Pruessner, Champagne, Meaney, & Dagher, Reference Pruessner, Champagne, Meaney and Dagher2004; Wand et al., Reference Wand, Oswald, McCaul, Wong, Johnson and Zhou2007), patients with schizophrenia, CHR, and first-degree relatives (Brunelin et al., Reference Brunelin, Amato, van Os, Costes, Suaud Chagny and Saoud2010; Lataster et al., Reference Lataster, Collip, Ceccarini, Hernaus, Haas and Booij2014; Mizrahi et al., Reference Mizrahi, Addington, Rusjan, Suridjan, Ng and Boileau2012).
Social deafferentation may predispose to plastic brain reorganization, leading to psychosis
Observations of social withdrawal long preceding psychotic symptoms, in the premorbid and prodromal phases of schizophrenia, is consistent with Hoffman's model (Reference Hoffman2007, Reference Hoffman2008), discussed earlier, which posits that the plastic brain reorganizes neural pathways following isolation to “produce spurious social meaning … in the form of complex, emotionally compelling hallucinations and delusions” (Hoffman, Reference Hoffman2008) Social deafferentation suggests a chain of causation, beginning with the effect of environment on neurobiology, followed by the impact of neurobiological changes and plasticity on experience. This concept also identifies a critical period during which both social withdrawal and deafferentation might result in psychotic symptoms. Initial, although only preliminary, evidence for the hypothesis has been obtained in CHR patients (Hoffman, Reference Hoffman2007).
Summary
The aberrant plasticity model and critical period concepts, as they relate to the premorbid and onset periods prior to psychosis, allow for the suggestion of an evolution of risk states that brings together various hypotheses of reduced plasticity predisposing to aberrant plastic reorganization of neural circuits. Early brain insults due to prenatal or early life adversity may lead to reduced cortical glutamatergic function and impaired experience-dependent neuroplasticity. This fits with the picture of early cognitive and learning deficits, and social withdrawal and deafferentation seen in premorbid studies of adolescents at risk for schizophrenia. In turn, reduced glutamatergic tone would result in decreased synaptic and gray matter density by the time of early adolescence. Combined with increased exposure to stressful situations and decreased cognitive adaptive capacity to them, these alterations would lead to a maladaptive plasticity cascade, that is, overactivation of the HPA axis (even beyond the normal HPA changes expected during adolescence) and dopaminergic stress responses that underlie affective dysregulation, risk for substance abuse, and eventually psychosis. This integrative pathophysiologic model might explain the “classic” trajectory of phenomena and symptomatology in high-risk populations.
Harnessing Neuroplasticity for Therapeutic (and Prophylactic) Gains
It is clear that observations of diminished as well as aberrant excessive plasticity motivate novel therapeutic as well as prophylactic therapeutic strategies in schizophrenia and the at-risk states for this illness. We herein provide examples of such emerging approaches.
Cognitive training
Cognitive training or cognitive remediation is an evolving form of intervention that allows us to intentionally harness neuroplastic processes related to learning for therapeutic purposes that target the disabling cognitive deficits of schizophrenia (Keshavan, Vinogradov, Rumsey, Sherill, & Wagner, Reference Keshavan, Vinogradov, Rumsey, Sherrill and Wagner2014). A recent meta-analysis suggested that cognitive training resulted in modest gains on cognition and socio-occupational functioning with mean effect sizes of 0.45 and 0.42, respectively (Wykes, Huddy, Cellard, McGurk, & Czobor, Reference Wykes, Huddy, Cellard, McGurk and Czobor2011). Besides, the benefits of some of these interventions are likely to last beyond treatment cessation (Eack, Greenwald, Hogarty, & Keshavan, Reference Eack, Greenwald, Hogarty and Keshavan2010; Subramaniam et al., Reference Subramaniam, Luks, Fisher, Simpson, Nagarajan and Vinogradov2012; Wykes et al., Reference Wykes, Reeder, Williams, Corner, Rice and Everitt2003).
The underlying plastic changes with cognitive training have been explored by neuroimaging studies. Patients who received cognitive training showed less gray matter loss in the left parahippocampal and fusiform gyrus and greater gray matter increases in the left amygdala after 2 years of cognitive enhancement therapy, as compared to a nonspecific supportive therapy (Eack, Hogarty, et al., Reference Eack, Hogarty, Cho, Prasad, Greenwald and Hogarty2010). It is interesting that patients with larger cortical thickness at baseline (higher cortical “reserve”) improved faster (Keshavan, Eack, et al., Reference Keshavan, Eack, Wojtalik, Prasad, Francis and Bhojraj2011). Computer-based cognitive training (a reality-monitoring task) has been shown to normalize the task-based activation of the prefrontal regions (Subramaniam et al., Reference Subramaniam, Luks, Fisher, Simpson, Nagarajan and Vinogradov2012), emotional-task based neural activations in the postcentral gyrus (Hooker et al., Reference Hooker, Bruce, Fisher, Verosky, Miyakawa and Vinogradov2012), and attention/executive task based activations of the dorsolateral prefrontal cortex, anterior cingulate, and frontopolar cortex (Haut, Lim & MacDonald, Reference Haut, Lim and MacDonald, III2010). In addition, specific training of auditory discrimination and verbal memory and not a broadly administered cognitive training showed normalization of abnormally reduced sensory gating in schizophrenia patients as measured using magnetoencephalography (Popov et al., Reference Popov, Jordanov, Rockstroh, Elbert, Merzenich and Miller2011). Diffusion tensor imaging studies provide additional evidence by revealing normalization of the interhemispheric connectivity between the bilateral prefrontal cortices via the corpus callosum in patients who received cognitive remediation (Penades et al., Reference Penades, Pujol, Catalan, Massana, Rametti and Garcia-Rizo2013). While structural and functional cortical plasticity changes have been demonstrated with cognitive training, one study also showed an increase in serum BDNF levels (Vinogradov et al., Reference Vinogradov, Fisher, Holland, Shelly, Wolkowitz and Mellon2009).
Brain stimulation approaches to improve symptoms by targeting cortical plasticity
Noninvasive brain stimulation strategies have been increasingly used to target specific regions of the brain, guided by existing neurobiological evidence of impaired (either reduced or excessive) activity in specific neural systems (Hasan, Wobrock, Rajji, Malchow, & Daskalakis, Reference Hasan, Wobrock, Rajji, Malchow and Daskalakis2013; Rajji, Rogasch, Daskalakis, & Fitzgerald, Reference Rajji, Rogasch, Daskalakis and Fitzgerald2013). Two symptom dimensions that have been commonly studied are negative symptoms, where high-frequency TMS pulses are applied to activate the left dorsolateral prefrontal cortex, and auditory hallucinations, where low-frequency TMS pulses are applied to inhibit the left temporoparietal cortex (Hoffman et al., Reference Hoffman, Boutros, Berman, Roessler, Belger and Krystal1999). In a meta-analysis of studies on high-frequency rTMS delivered to the left dorsolateral prefrontal cortex, it was shown that the rTMS improved negative symptoms of schizophrenia with a modest effect size of 0.43 (Dlabac-de Lange, Knegtering, & Aleman, Reference Dlabac-de Lange, Knegtering and Aleman2010). Similar findings were also replicated in a larger, more recent meta-analysis (Shi, Yu, Cheung, Shum, & Chan, Reference Shi, Yu, Cheung, Shum and Chan2014). This is still an evolving treatment modality, and one of the means to optimize the therapeutic benefit is by targeting different sites like deeper prefrontal cortices (Levkovitz, Rabany, Harel, & Zangen, Reference Levkovitz, Rabany, Harel and Zangen2011) or even the cerebellar vermis (Demirtas-Tatlidede et al., Reference Demirtas-Tatlidede, Freitas, Cromer, Safar, Ongur and Stone2010).
A recent meta-analysis of five randomized, double blind, sham-controlled studies reported that low-frequency rTMS delivered to the left temporoparietal cortex improved auditory hallucinations with a modest effect size of 0.44 (Slotema, Aleman, Daskalakis, & Sommer, Reference Slotema, Aleman, Daskalakis and Sommer2012). Furthermore, another study using the same investigation demonstrated a reduction in cerebral blood flow in the primary auditory cortex, left Broca's area, and cingulate gyrus in patients who received 10-day rTMS sessions for auditory hallucinations (Kindler et al., Reference Kindler, Homan, Jann, Federspiel, Flury and Hauf2013). Recently, tDCS has been shown to be beneficial in treating medication-resistant auditory hallucinations in schizophrenia (Brunelin et al., Reference Brunelin, Mondino, Gassab, Haesebaert, Gaha and Suaud-Chagny2012).
A third application of brain stimulation in schizophrenia that is gaining preliminary empirical support is in treating cognitive deficits (Guse, Falkai, & Wobrock, Reference Guse, Falkai and Wobrock2010). A single session of 20-Hz rTMS delivered to the bilateral dorsolateral prefrontal cortex resulted in a potentiation of the frontal gamma oscillatory activity during a working-memory task in healthy individuals (Barr et al., Reference Barr, Farzan, Rusjan, Chen, Fitzgerald and Daskalakis2009). This sequence of bilateral rTMS administered in schizophrenia patients for 4 weeks, was compared to stimulation using sham rTMS in a randomized controlled trial. It was found that the group receiving true rTMS performed significantly better on a working-memory task at the end of the 4-week trial (Barr et al., Reference Barr, Farzan, Rajji, Voineskos, Blumberger and Arenovich2013). However, another study using unilateral (left) 10-Hz rTMS did not find similar benefits (Guse et al., Reference Guse, Falkai, Gruber, Whalley, Gibson and Hasan2013). Application of rTMS for cognitive enhancement is still in its infancy and requires more studies to standardize and optimize the treatment protocols. One way ahead may be to target modulation of mirror neuron regions to enhance social cognitive performance (Mehta, Thirthalli, et al., Reference Mehta, Thirthalli, Basavaraju, Gangadhar and Pascual-Leone2013).
Medications
Antipsychotic medications are the most common form of therapeutic intervention in schizophrenia. Multiple studies have demonstrated that antipsychotic medications induce both anatomical- and molecular-level neuroplastic changes in the brain (Konradi & Heckers, Reference Konradi and Heckers2001). Studies using rat hippocampal neuronal cultures have revealed adaptive changes in postsynaptic density proteins, dendritic spine morphology, BDNF expression, and excitatory postsynaptic potentials (Critchlow, Maycox, Skepper, & Krylova, Reference Critchlow, Maycox, Skepper and Krylova2006; Pandya, Kutiyanawalla, & Pillai, Reference Pandya, Kutiyanawalla and Pillai2013; Park et al., Reference Park, Lee, Cho, Seo, Lee and Lee2013; Shim, Hammonds, Tatsuoka, & Feng, Reference Shim, Hammonds, Tatsuoka and Feng2012). It is interesting that typical and atypical antipsychotics have a differential regulation of synaptic plasticity by modulating activity of different postsynaptic proteins (Critchlow et al., Reference Critchlow, Maycox, Skepper and Krylova2006). At a molecular level, typical antipsychotic medication-induced plasticity changes are largely observed in the striatum and nucleus accumbens, whereas atypical antipsychotic drugs have a subtler and more widespread impact (Konradi & Heckers, Reference Konradi and Heckers2001).
Such a pattern is partially corroborated by structural neuroimaging studies. Treatment with typical antipsychotic medications is associated with enlargement of the striatum and other structures in the basal ganglia and reduction in frontal, temporal, and parietal cortical gray matter volume (Dazzan et al., Reference Dazzan, Morgan, Orr, Hutchinson, Chitnis and Suckling2005; Lieberman et al., Reference Lieberman, Tollefson, Charles, Zipursky, Sharma and Kahn2005; Smieskova et al., Reference Smieskova, Fusar-Poli, Allen, Bendfeldt, Stieglitz and Drewe2009). Atypical antipsychotic medications are associated with enlargement of thalamus and cortical gray matter volumes (Dazzan et al., Reference Dazzan, Morgan, Orr, Hutchinson, Chitnis and Suckling2005; Deng et al., Reference Deng, McAlonan, Cheung, Chiu, Law and Cheung2009; Scherk & Falkai, Reference Scherk and Falkai2006), as well as, reduction in other cortical (medial frontal gyrus) volumes (Deng et al., Reference Deng, McAlonan, Cheung, Chiu, Law and Cheung2009). It is intriguing that potential neuroplastic changes are seen considerably early. A pharmacological-MRI investigation in humans reported striatal volume changes and structural–functional decoupling in motor circuits within hours of administering D2-receptor blockers (Tost et al., Reference Tost, Braus, Hakimi, Ruf, Vollmert and Hohn2010). It is however important to note that it is still unclear as to whether long-term cortical volume change is a function of antipsychotic medications or of disease progression (Andreasen, Liu, Ziebell, Vora, & Ho, Reference Andreasen, Liu, Ziebell, Vora and Ho2013).
The therapeutic efficacy of lithium may also be explained based on its ability to modulate synaptic plasticity. Chronic lithium treatment increases dendritic branching in hippocampal neurons, and also enhances LTP-like plasticity (Shim et al., Reference Shim, Hammonds, Tatsuoka and Feng2012). Lithium in humans can cause a switch from LTD- to LTP-like plasticity using TMS (Voytovych, Krivanekova, & Ziemann, Reference Voytovych, Krivanekova and Ziemann2012). Lithium's effects on BDNF (Voytovych et al., Reference Voytovych, Krivanekova and Ziemann2012; Yasuda, Liang, Marinova, Yahyavi, & Chuang, Reference Yasuda, Liang, Marinova, Yahyavi and Chuang2009) and on the B-cell lymphoma 2 (BCL2) family of genes that regulate apoptosis (Beech et al., Reference Beech, Leffert, Lin, Sylvia, Umlauf and Mane2014; Lowthert et al., Reference Lowthert, Leffert, Lin, Umlauf, Maloney and Muralidharan2012) may explain these observed effects.
Erythropoietin has important neurotrophic and immunomodulatory functions (Rabie & Marti, Reference Rabie and Marti2008) and has shown improvement in cognitive performance in a controlled trial in schizophrenia (Ehrenreich et al., Reference Ehrenreich, Hinze-Selch, Stawicki, Aust, Knolle-Veentjer and Wilms2007). Overall, these novel treatment strategies provide broader therapeutic avenues for clinicians to harness neuroplasticity in aiding patients with schizophrenia.
Other approaches
Physical exercise (wheel running) in mice has shown to increase neurogenesis, dendritic proliferation, and LTP in the dentate gyrus of the hippocampus (van Praag, Kempermann, & Gage, Reference van Praag, Kempermannx and Gage1999). In humans, regular aerobic exercise increases hippocampal and cortical volumes and improves cognitive performance in the elderly (Erickson et al., Reference Erickson, Voss, Prakash, Basak, Szabo and Chaddock2011), as well as in early middle adulthood (Killgore, Olson, & Weber, Reference Killgore, Olson and Weber2013). Plausible mechanisms include increased blood flow and oxygenation to the hippocampus (Pereira et al., Reference Pereira, Huddleston, Brickman, Sosunov, Hen and McKhann2007) and greater production of BDNF (Vaynman, Ying, & Gomez-Pinilla, Reference Vaynman, Ying and Gomez-Pinilla2004). Pajonk et al. (Reference Pajonk, Wobrock, Gruber, Scherk, Berner and Kaizl2010) have extended this work in patients with chronic schizophrenia. They found that 3 months of aerobic exercise, as opposed to control condition (playing table football), not only increased hippocampal volumes but also resulted in greater N-acetylaspartate to creatine ratio in the hippocampus, and improved short-term memory of these patients.
Other novel therapeutic options that can enhance synaptic plasticity include enriched environment. Providing an enriched environment comprising novel and complex sensory, cognitive, social, and motor stimuli can boost key neural circuits to bring about adaptive behavioral change. This has been demonstrated in rodents, where enriched environments have resulted in neuroplasticity-driven molecular, cellular, and behavioral changes. These plasticity-harnessing benefits have been demonstrated in rodent models of Alzheimer dementia, schizophrenia, and autism spectrum disorders (Hannan, Reference Hannan2014; Pang & Hannan, Reference Pang and Hannan2013). Integrating specific dimensions of environmental enrichment in rehabilitation programs for schizophrenia patients needs further study.
Conclusion
We have outlined evidence that the core manifestations of schizophrenia (positive, negative, and cognitive symptoms) may be understood as resulting from aberrant neuroplasticity. As shown in patients with schizophrenia as well as at-risk populations, this dysplasticity may involve both hypoplasticity in key brain systems serving cognitive functions and goal-directed behavior, and hyperplasticity in neural systems governing salience detection, emotion processing, and regulation as well as default mode systems. It is possible that the hyperplasticity is a maladaptive response in an effort to compensate for a primary impairment in plasticity, though the converse is also possible (Figure 3). Preventive and therapeutic interventions with medications, neuromodulation, and psychosocial treatments may be directed both at reversing plasticity deficits as well as harnessing compensatory neuroplasticity in more adaptive channels. The conceptual model we have developed herein has heuristic value, and suggests several testable questions for future research. The increasing understanding of brain mechanisms underlying plasticity may suggest new ways of detecting preclinical disease, better biomarkers to guide treatment selection, and novel therapeutic targets.
A number of essential questions remain to be examined and answered by future generations of basic and clinical research: (a) What specific mechanisms of altered plasticity contribute to schizophrenia? Are they primary rather than secondary to other pathophysiological processes, and what gene–environment interactions underlie such mechanisms? (b) What underlies the variable trajectories among high-risk individuals, for example, between those who to convert to psychosis versus nonconverters, and (in nonconverters) between those who develop cognitive versus affective, anxiety, or substance misuse disorders? (c) Among proband groups, do differences in plasticity capacities predict differential outcome trajectories? (d) What are the best ways to noninvasively harness plasticity in the service of prevention and early intervention? (e) Are the plasticity alterations unique to specific diagnoses, or do they vary dimensionally across a broad spectrum of major psychiatric disorders? (f) Can critical windows of neuroplasticity be reopened in patients who are already on a trajectory to major mental illness or who have an already-evident disorder?




