


Externalizing disorders, such as substance use disorders and adult antisocial personality disorder, are common (British Psychological Society, 2010; Esser et al., Reference Esser, Hedden, Kanny, Brewer, Gfroerer and Naimi2014; Grant et al., Reference Grant, Stinson, Dawson, Chou, Dufour and Compton2006; Substance Abuse and Mental Health Administration, 2013) and associated with a myriad of poor psychosocial and physical health outcomes (British Psychological Society, 2010; Centers for Disease and Control, 2014; Grieg, Baker, Lewin, Webster, & Carr, Reference Greig, Baker, Lewin, Webster and Carr2006). Externalizing disorders are complex in that they entail a dynamic interplay of both heritable and environmental influences (Hicks, Foster, Iacono, & McGue, Reference Hicks, Foster, Iacono and McGue2013; Rhee & Waldman, Reference Rhee and Waldman2002). Two processes that are essential to delineating the mechanisms underlying externalizing disorders are gene–environment correlation and interaction. Gene–environment correlation refers to the process whereby genetically influenced traits are associated with exposure to environmental risk (Scarr & McCartney, Reference Scarr and McCartney1983). Gene × Environment interaction refers to the process wherein genetic influences vary as a function of environmental context (Dick, Reference Dick2011; Rutter, Moffitt, & Caspi, Reference Rutter, Moffitt and Caspi2006).Footnote 1
Both gene–environment correlation and interaction have long been shown to contribute to adolescent externalizing problems (i.e., antisocial behavior and problematic substance use; Benner, Kretsch, Harden, & Crosnoe, Reference Benner, Kretsch, Harden and Crosnoe2014; Button, Lau, Maughan, & Eley, Reference Button, Lau, Maughan and Eley2008; Byrd & Manuck, Reference Byrd and Manuck2014; Cicchetti, Rogosch, & Thibodeau, Reference Cicchetti, Rogosch and Thibodeau2012; Cleveland, Wiebe, & Rowe, Reference Cleveland, Wiebe and Rowe2005; Dick et al., Reference Dick, Pagan, Viken, Purcell, Kaprio and Pulkkinen2007; Feinberg, Button, Neiderhiser, Reiss, & Hetherington, Reference Feinberg, Button, Neiderhiser, Reiss and Hetherington2007; Hicks, South, Dirago, Iacono, & McGue, Reference Hicks, South, Dirago, Iacono and McGue2009). Few studies, however, have examined whether these processes are also present in young adulthood, or whether exposure to environmental risk factors in adolescence has long-lasting effects in terms of moderating genetic risk for externalizing disorders in adulthood. This is a critical gap to fill because it remains imperative to understand how both early and later developmental context may affect the etiology of externalizing disorders across time.
Gene–Environment Interaction and Correlation in Adolescence
The ubiquity of gene–environment correlation processes in adolescence was demonstrated by a landmark meta-analysis that showed many variables assumed to be “environmental” (e.g., parenting and peer interaction) are substantially influenced by additive genetic factors (Kendler & Baker, Reference Kendler and Baker2007). Furthermore, research has consistently shown that the associations between such environmental factors and externalizing disorders are at least partially attributable to common additive genetic influence (e.g., Marceau et al., Reference Marceau, Horwitz, Narusyte, Ganiban, Spotts and Reiss2013). These findings are often interpreted as supporting the notion of gene–environment correlation (Scarr & McCartney, Reference Scarr and McCartney1983), or how exposure and selection into specific environmental contexts are related to our unique genotypes. One such process is evocative gene–environment correlation, or how children and adolescents evoke certain types of responses from parents and others based on their genetically influenced traits (e.g., some children may evoke frustration or negativity whereas others may evoke warmth or positivity based on their genetically influenced temperament). An alternative process is active gene–environment correlation, or how children and adolescents actively select out a particular environment based on their unique genotype (e.g., children likely seek out affiliation with like-minded antisocial or prosocial friends).
Potentially as crucial is that once people have been exposed or selected into specific environmental contexts, such contexts also appear to either amplify or offset genetic risk for externalizing disorders, a process known as Gene × Environment interaction. For example, Hicks (Reference Hicks, South, Dirago, Iacono and McGue2009) showed a consistent pattern of Gene × Environment interaction for externalizing disorders and several environmental risk factors in adolescence. Specifically, environmental factors that included antisocial peer affiliation, parent–child relationship problems, academic engagement, and stressful life events all moderated additive genetic influences on externalizing disorders such that genetic influences were greater in the context of greater environmental adversity. Because of the consistency of the findings across environmental variables, Hicks et al. speculated that the pattern of increased genetic risk as a function of greater environmental adversity may be a general mechanism underlying risk for externalizing disorders.
Gene × Environment × Development Interplay?
Although research has clearly supported the notion of gene–environment correlation and interaction involving key family, peer, and school factors in relation to adolescent externalizing disorders (see citations above), less research has examined the constancy of gene–environment interplay beyond adolescence and into young adulthood when key externalizing attributes are more common. Specifically, antisocial behavior tends to peak in late adolescence (Blumenstein, Cohen, & Farrington, Reference Blumenstein, Cohen and Farrington1988; Loeber et al., Reference Loeber, Menting, Lynam, Moffitt, Stouthamer-Loeber and Stallings2012; Moffitt, Reference Moffitt1993), and heavy substance use and substance use disorders tend to peak in the early 20s (Centers for Disease and Control, 2012; Johnston, O'Malley, Bachman, & Schulenberg, Reference Johnston, O'Malley, Bachman and Schulenberg2009; Schulenberg & Maggs, Reference Schulenberg and Maggs2002; Substance Abuse and Mental Health Administration, 2014). It may be that gene–environment interaction is more relevant to the development of externalizing disorders at the end of adolescence or in early adulthood when they are most common. Alternatively, it may be that gene–environment interaction may be more relevant earlier in development when individuals may be more malleable or sensitive to environmental context (Kendler, Gardner, & Dick, Reference Kendler, Gardner and Dick2011).
Converging evidence supports the latter notion in that gene–environment interaction involving externalizing disorders may be developmentally limited to adolescence. For example, follow-up analyses using the same sample as Hicks et al. (Reference Hicks, South, Dirago, Iacono and McGue2009) found that poor parent–child relationship quality (Samek, Hicks, et al., Reference Samek, Hicks, Keyes, Bailey, McGue and Iacono2015) and low academic achievement (Johnson, McGue, & Iacono, Reference Johnson, McGue and Iacono2009) did not moderate genetic risk for externalizing disorders at age 24. Instead, gene–environment correlation explained much of the concurrent and long-term associations between parent–child relationship quality and young adult externalizing disorders (Samek, Hicks, et al., Reference Samek, Hicks, Keyes, Bailey, McGue and Iacono2015), and shared environmental influences explained much of the association between higher educational attainment and young adult's antisocial behavior symptoms (Johnson et al., Reference Johnson, McGue and Iacono2009). Thus, other confounding family factors (genetic and environmental) explain much of the long-term association between these adolescent environmental contexts and adult outcomes rather than Gene × Environment interaction processes.
However, it remains unclear whether other aspects of young adult's environmental context may amplify or offset genetic risk for externalizing disorders: particularly those aspects that may be more important to young adults than parenting or school. Because antisocial peer affiliation is the one of the strongest environmental correlates of externalizing problems across childhood, adolescence, and young adulthood (Brendgen, Reference Brendgen2012; Dishion & Owen, Reference Dishion and Owen2002; Lansford, Yu, Pettit, Bates, & Dodge, Reference Lansford, Yu, Pettit, Bates and Dodge2014; Monahan, Rhew, Hawkins, & Brown, Reference Monahan, Rhew, Hawkins and Brown2014; Monahan, Steinberg, & Cauffman, Reference Monahan, Steinberg and Cauffman2009; Wichers, Gillespie, & Kendler, Reference Wichers, Gillespie and Kendler2013), it may continue to be a factor that amplifies or offsets genetic risk for externalizing disorders in early adulthood.
Antisocial Peer Affiliation
A large body of research has demonstrated important bidirectional influence of peers and substance use in adolescence (Curran, Stice, & Chasin, Reference Curran, Stice and Chassin1997; Van Ryzin & Dishion, Reference Van Ryzin and Dishion2014; Van Ryzin, Fosco, & Dishion, Reference Van Ryzin, Fosco and Dishion2012); however, comparatively fewer investigations have evaluated such effects in early adulthood. In a landmark study, Dishion and Owen (Reference Dishion and Owen2002) showed evidence of both selection and socialization in the linkages between substance use and deviance within friendships. Specifically, Dishion and Owen showed that affiliation with substance-using friends in early adolescence was associated with subsequent substance use in middle adolescence, which then reinforced a pattern of associating with deviant friends in late adolescence and subsequent substance use in early adulthood. In relation to antisocial behavior more generally, other research suggests little impact of peer socialization beyond age 20 (Monahan et al., Reference Monahan, Steinberg and Cauffman2009), particularly in relation to an aggregate externalizing measure (Samek, Goodman, Erath, McGue, & Iacono, Reference Samek, Goodman, Erath, McGue and Iaconoin press). Thus, it may be that gene–environment interaction involving antisocial peer affiliation and externalizing disorders is less relevant in adulthood than in adolescence.
Prior research has shown that child and adolescent antisocial peer affiliation is heritable (Bullock, Deater-Deckard, & Leve, Reference Bullock, Deater-Deckard and Leve2006; Cleveland et al., Reference Cleveland, Wiebe and Rowe2005) and that the association between adolescent antisocial peer affiliation and adolescent externalizing problems is largely explained by shared genetic factors (Fowler et al., Reference Fowler, Shelton, Lifford, Rice, McBride and Nikolov2007; Teneyck & Barnes, Reference Teneyck and Barnes2015). Furthermore, Kendler et al. (Reference Kendler, Jacobson, Gardner, Gillespie, Aggen and Prescott2007) showed heritability estimates of retrospective reports of antisocial peer affiliation increased and then stabilized over time. Specifically, the heritability of antisocial peer affiliation increased from about 30% for 8- to 11-year-olds to about 50% for ages 15–17, 18–21, and 22–25. One possibility is that as antisocial peer affiliation becomes more heritable over time, gene–environment correlation processes become even more important to the emergence and maintenance of externalizing disorders, but this hypothesis has not yet been evaluated.
Prior research has also consistently shown a Gene × Environment interaction such that the additive genetic influences on adolescent externalizing problems are greater in the context of greater antisocial peer affiliation (Fowler et al., Reference Fowler, Shelton, Lifford, Rice, McBride and Nikolov2007; Harden, Hill, Turkheimer, & Emery, Reference Harden, Hill, Turkheimer and Emery2008; Hicks et al., Reference Hicks, South, Dirago, Iacono and McGue2009). It is unknown, however, if antisocial peer affiliation continues to amplify genetic risk for externalizing disorders in young adulthood or if this Gene × Environment interaction is developmentally limited to adolescence as has been found for parenting and school factors (Johnson et al., Reference Johnson, McGue and Iacono2009; Samek, Hicks, et al., Reference Samek, Hicks, Keyes, Bailey, McGue and Iacono2015). An effect of antisocial peer affiliation limited to adolescence would be consistent with several nongenetically informed studies demonstrating that individuals become more resistant to antisocial peer influences over time (Gardner & Steinberg, Reference Gardner and Steinberg2005; Sumter, Bokhorst, Steinberg, & Westenberg, Reference Sumter, Bokhorst, Steinberg and Westenberg2009), potentially due to continued brain development and improvement in cognitive skills throughout adolescence and young adulthood (Albert, Chein, & Steinberg, Reference Albert, Chein and Steinberg2013).
Failure to detect a Gene × Environment interaction between antisocial peer affiliation and adult externalizing disorders, combined with similar findings for parenting and academic achievement (Johnson et al., Reference Johnson, McGue and Iacono2009; Samek, Hicks, et al., Reference Samek, Hicks, Keyes, Bailey, McGue and Iacono2015), would suggest that adolescence may be a critical period wherein greater autonomy and exposure into high-risk environments provides a catalyst for previously unexpressed genetic risk. Once initiated, however, selection or active gene–environment correlation processes may maintain the association between externalizing disorders and contextual risk in adulthood.
We tested this research question by examining gene–environment interplay between externalizing disorders and antisocial peer affiliation at multiple time points spanning adolescence (age 17), early adulthood (ages 20 and 24), and later adulthood (age 29). Further, we tested whether adolescent antisocial peer affiliation had moderating effects on genetic influences on adult externalizing disorders. Given prior research (Johnson et al., Reference Johnson, McGue and Iacono2009; Monahan et al., Reference Monahan, Steinberg and Cauffman2009; Samek, Hicks, et al., Reference Samek, Hicks, Keyes, Bailey, McGue and Iacono2015), as well as our theory that adolescence may be a developmentally limited critical period wherein high-risk environments provide a catalyst for previously unexpressed genetic risk, we predicted that antisocial peer affiliation would moderate additive genetic influences on externalizing disorders in adolescence. We also expected that adolescent antisocial peer affiliation would not moderate additive genetic influences on adult externalizing disorders. Rather, we predicted that externalizing disorders and antisocial affiliation would exhibit significant additive genetic correlations (i.e., common genetic influences), both concurrently and prospectively across adolescence and young adulthood (thus evidencing greater selection or active gene–environment correlation than gene–environment interaction mechanisms postadolescence).
We evaluated these hypotheses using an aggregated count of symptoms of clinical externalizing disorders, including several substance use disorders and the adult criteria for antisocial personality disorder (hereafter referred to as adult antisocial behavior symptoms). However, given the slight difference in age at which rates of antisocial behavior versus substance use disorder peak (e.g., see Loeber et al., Reference Loeber, Menting, Lynam, Moffitt, Stouthamer-Loeber and Stallings2012; Moffitt, Reference Moffitt1993; Substance Abuse and Mental Health Administration, 2014), we also evaluated the extent of differences in gene–environmental interplay involving antisocial behavior versus substance use disorder by developmental stage.
Method
Participants were members of the Minnesota Twin Family Study (Iacono, Carlson, Taylor, Elkins, & McGue, Reference Iacono, Carlson, Taylor, Elkins and McGue1999), a longitudinal, cohort-sequential study of twins born in Minnesota. Twin families were identified using publicly available birth certificates (birth years 1972 to 1984) and were located using several public databases. Participating families had twins who were the biological offspring of their parents and lived within a day's drive of the university laboratory; neither twin could have a mental or physical handicap that would impair study participation. About 90% of twins were successfully located, and 83% of eligible and located families agreed to participate. After a description of the study to twins and parents, parents provided written consent for minors and minors provided written assent, while those 18 years and older provided written consent. The University of Minnesota Institutional Review Board approved all study protocols.
The sample used both the older and younger cohort of twins, thus included 2,764 individuals from 1,382 same-sex twin pairs (52% female, 65% monozygotic), assessed at the target ages of 17 (M = 17.8 years, SD = 0.69, N = 2,577), 20 (M = 21.0 years, SD = 0.82, N = 2,450), 24 (M = 25.0 years, SD = 0.90, N = 2,499), and 29 (M = 29.4 years, SD = 0.67, N = 2,496) years old (see Iacono et al., Reference Iacono, Carlson, Taylor, Elkins and McGue1999, for a detailed study overview). Consistent with demographics of Minnesota for the relevant birth years, nearly all participants were of European American ancestry (95%). Participation rates ranged from 88% to 93% across follow-up assessments. To evaluate attrition, we compared mean differences in symptoms of externalizing disorder symptoms at age 17 for those who did or did not complete adult assessments. Those who participated in adult assessments had slightly fewer externalizing symptoms at age 17 than those who did not participate, but the effect sizes were small with mean Cohen d values of –0.21, –0.16, and –0.03 at ages 20, 24, and 29, respectively, indicating little evidence of attrition effects.
Measures
Antisocial peer affiliation
Participants rated characteristics of their peer groups using the Friends survey (Burt, McGue, & Iacono, Reference Burt, McGue and Iacono2009; Walden, McGue, Lacono, Burt, & Elkins, Reference Walden, McGue, Iacono, Burt and Elkins2004), which consisted of 7 to 9 items depending on developmental stage.Footnote 2 Items (e.g., “my friends enjoy getting drunk” and “my friends know where to buy drugs”) were rated from 1 (all of my friends are like that) to 4 (none of my friends are like that); αs ranged from 0.83 to 0.88.
Externalizing disorders
Structured interviews were used to assess DSM-III-R symptoms of nicotine dependence, alcohol use disorder, illicit drug use disorder, and adult antisocial behavior (adult criteria for antisocial personality disorder) at ages 17, 20, 24, and 29. Substance use disorders were assessed using the Substance Abuse Module of the Composite International Diagnostic Interview (Robins, Babor, & Cottler, Reference Robins, Babor and Cottler1987), and adult antisocial behavior was assessed using an interview adapted from the Structured Clinical Interview for DSM-III-R Axis II (Spitzer, Williams, & Gibbon, Reference Spitzer, Williams and Gibbon1987). Interviews were reviewed by at least two individuals with advanced training in clinical diagnoses, and consensus among the reviewers was reached prior to symptom assignment. Reliability was assessed by double-coding a randomly selected subsample of 600 Minnesota Twin Family Study participants. Kappa coefficients indexing diagnostic reliability were >0.90 for all substance use disorders and 0.79 for adult antisocial behavior. Abuse and dependence symptoms were combined to calculate symptom count variables for alcohol and illicit drug disorders. Because we were interested in evaluating a general measure of externalizing disorders, symptom counts were standardized (z scored) and averaged to calculate an externalizing composite at each age (mean r values among z score scales were .50 at age 17, .45 at age 20, and .39 at ages 24 and 29). Subsequent analyses evaluated differences in antisocial behavior versus substance use disorders. Here, substance use disorder symptom counts were standardized and averaged within each age (mean r values among z score scales were .53 at age 17, .41 at age 20, .34 at age 24, and .35 at age 29).
Analytic plan
Structural equation modeling was used to evaluate gene–environment interplay for the concurrent and prospective associations between antisocial peer affiliation and externalizing disorders using Mx software (Neale, Reference Neale2006). Full information maximum likelihood was used to adjust parameter estimates for missing data (Enders & Bandalos, Reference Enders and Bandalos2001; Johnson & Young, Reference Johnson and Young2011). To better approximate normality, externalizing composites were log-transformed prior to analysis. Consistent with prior research, sex, age, age2, and Age × Sex were covaried out of all phenotypes prior to modeling (age and sex adjustments were conducted within assessment). Univariate and bivariate models were first fit to estimate the additive genetic (A), shared environmental (C), and nonshared environmental (E) influences on study phenotypes, where A refers to genetic influences on twin similarity, C refers to environmental influences on twin similarity, and E refers to environmental influences on twin differences. ACE parameters are estimated by comparing the relative similarity of monozygotic (MZ) and dizygotic (DZ) twin pairs. Additive genetic effects are inferred when MZ correlations are greater than DZ correlations. Shared environmental effects are inferred when DZ correlations are greater than half of MZ correlations. Nonshared environmental effects are inferred when MZ correlations are less than 1.0 (for a detailed overview of ACE modeling, see Rijsdijk & Sham, Reference Rijsdijk and Sham2002).
Next, we tested for Gene × Environment interaction in the presence of gene–environment correlation (Purcell, Reference Purcell2002). As illustrated in Figure 1, this bivariate analysis decomposes the ACE contributions on the covariance between antisocial peer affiliation and externalizing disorders (a11*a21, c11*c21, e11*e21) and the variance unique to externalizing disorders (a22, c22, e22). In this bivariate decomposition, the genetic and environmental covariance (e.g., a11*a21) is standardized to give estimates of genetic and environmental correlations (range –1.0 to 1.0). As shown in Figure 1, in the case of a Gene × Environment interaction, ACE parameters are also adjusted for the direction and size of the moderation (β) and the level of the moderator (M), here antisocial peer affiliation. Moderation can occur on ACE effects common to antisocial peer affiliation and externalizing disorders (a21 + βa21 × M, c21 + βc21 × M, e21 + βe21 × M) or unique to externalizing disorders (a22 + βa22 × M, c22 + βc22 × M, e22 + βe22 × M). Model fit was evaluated using the –2 log likelihood (–2LL) and testing the likelihood ratio test between comparison models. Several information theoretic fit indices were also used to evaluate fit including the Akaike information criterion, the sample-size adjusted Bayesian information criterion, and the deviance information criterion, in which smaller values indicated better fit. For all model comparisons, we compared the full ACE moderation model to a model that dropped all ACE moderation parameters. If the full ACE moderation model fit better than the no ACE moderation model, follow-up comparisons were made by dropping nonsignificant parameters (i.e., 95% confidence intervals that included zero) to identify the best fitting, most parsimonious model.
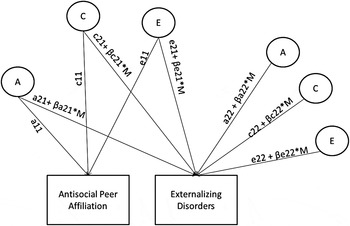
Figure 1. Gene–environment interaction in the presence of gene–environment correlation. Separate models were evaluated for antisocial peer affiliation in relation to externalizing disorders at ages 17, 20, 24, and 29, in addition to models evaluating the prospective relationship between antisocial peer affiliation at age 17 with externalizing disorders at ages 20, 24, and 29. A, Genetic influences; C, shared environmental influences; E, nonshared environmental influences. Parameters a11, c11, and e11 refer to the genetic and environmental influences on the moderator (antisocial peer affiliation). Parameters a21, c21, and e21 refer to the genetic and environmental influences on the moderator (antisocial peer affiliation) in common with the dependent variable (externalizing disorders). Parameters a22, c22, and e22 refer to the unique genetic and environmental influence on the dependent variable (externalizing disorders); β describes the magnitude and direction of moderation effect; and M indicates the level of the moderator. Moderation can influence both/either the common or unique variance for externalizing disorders.
Results
Preliminary analyses
Descriptive statistics and phenotypic correlations for study phenotypes are provided in Tables 1 and 2, respectively. Males had significantly higher mean externalizing symptom counts than females across all externalizing disorders (all ps < .05). All phenotypes were significantly and substantially correlated (mean r = .52, range = .37–.71, all ps < .001).
Table 1. Means (standard deviations) of externalizing symptom counts across sex

Note: The sample sizes reported for males and females reference the eligible sample size (total sample); however, specific numbers (ns) for each measure varied across assessment and by externalizing disorder (at age 17, ns = 1,191–1,246 for males and 1,336–1,369 for females; at age 20, ns = 1,103–1,112 for males and 1,326–1,336 for females; at age 24, ns = 1,130–1,172 for males and 1,296–1,317 for females; at age 29, ns = 1,181–1,182 for males and 1,314–1,314 for females). Nicotine dependence symptom counts ranged from 0 to 7. Alcohol and illicit drug use disorder symptom counts (abuse + dependence) ranged from 0 to 10. Adult antisocial behavior (adult criteria for adult antisocial personality disorder) symptom counts ranged from 0 to 10. Males had significantly higher mean symptom counts than females for all externalizing disorders (p < .05). The Cohen d values show the magnitude of the gender difference effect: 0.2–0.3 is considered small, 05 medium, and >0.8 large.
a This include five triplets that were not used in subsequent twin analyses.
Table 2. Phenotypic correlations

Note: EXT, Externalizing disorders. All variables were age, sex, Age × Age, and Age × Sex adjusted prior to phenotypic and biometric analysis. All correlations were significantly different from zero at p < .001.
Twin correlations and univariate ACE estimates are reported in Table 3. Additive genetic influences on the externalizing composites were moderate and stable over time (a 2 range = 0.46–0.56). Shared environmental influences on externalizing disorders were small at age 17 (c 2 = 0.16) and not significantly different from zero after age 17. Additive genetic influences on antisocial peer affiliation were small to moderate across time (a 2 range = 0.21–0.51). Shared environmental influences on antisocial peer affiliation were moderate through age 24 and not significantly different from zero at age 29. Nonshared environmental influence on externalizing disorders and antisocial peer affiliation tended to increase over time.
Table 3. Twin correlations and standardized ACE estimates

Note: EXT, Externalizing disorders. This table shows intraclass twin correlations for monozygotic (MZ) and dizygotic (DZ) twins as well as standardized estimates of additive genetic (a2 ), shared environmental (c2 ), and nonshared environmental (e2 ) influences from univariate decompositions.
a Sample sizes (n) shown for MZ and DZ are based on eligible sample size (total sample); actual numbers (ns) associated with correlations ranged from 681 to 823 for MZ pairs and 369 to 442 for DZ pairs (across measure and age of assessment). Ninety-five percent confidence intervals are provided in parentheses. Coefficients are significant if the confidence interval does not cross zero (nonsignificant coefficients are shown in bold italic for clarity of presentation). All variables were adjusted for age, sex, Age × Sex, and age2 prior to analysis.
Gene–environment correlations
Cross-sectional and longitudinal phenotypic, genetic, and environmental correlations between antisocial peer affiliation and externalizing composites are reported in Table 4. Genetic correlations between antisocial peer affiliation and the externalizing composite were medium to large (range = 0.47–0.86), and all were significantly different from zero (as indicated by the 95% confidence intervals not crossing zero). Shared environmental correlations were also large, though total shared environmental variance was small for the externalizing composites, suggesting little practical effect. Nonshared environmental correlations were moderate for the cross-sectional correlations at each time point and small for the longitudinal correlations (i.e., age 17 to 20, age 17 to 24, and age 17 to 29). Table 4 also shows the proportion of the phenotypic covariance due to genetic and environmental influence (columns add to 1.0). As expected given the genetic correlations, common genetic influences were substantial within and across time.
Table 4. Phenotypic, genetic, and environmental correlations between antisocial peer affiliation and externalizing disorders from ages 17 to 29

Note: EXT, Externalizing disorders. The table shows phenotypic correlations (r) as well as additive genetic (rA), shared environmental (rC), and nonshared environmental (rE) correlations from bivariate, full ACE Cholesky decompositions. It also shows the proportion of phenotypic covariance that is due to additive genetic (A), shared environmental (C), and nonshared environmental (E) influence, that is, how much genetic and environmental influences contributed to the total phenotypic covariation (r in column 1); note that the last three columns add to 1.0. For example, 36% of the total covariation between antisocial peer affiliation and EXT at age 17 was due to additive genetic influences, 50% due to shared environmental influences, and 14% due to nonshared environmental influences (36 + 50 + 14 = 100%). All variables were adjusted for age, sex, Age × Sex, and age2 prior to analysis. Significant coefficients are those with a confidence interval that does not cross zero (those that are not significant are denoted in bold italic for clarity of presentation).
Gene × Environment interaction in adolescence
Table 5 shows the fit statistics for all Gene × Environment interaction models. For the cross-sectional association between antisocial peer affiliation and the externalizing composite at age 17, as reported in rows 1 and 2, the no ACE moderation model fit significantly worse than the full ACE moderation model. This was evidenced by the significant likelihood ratio test and poorer fit in Akaike information criterion, adjusted the sample-size adjusted Bayesian information criterion, and deviance information criterion values. For the best fitting model (row 3), only parameters that were significantly different from zero (i.e., 95% confidence interval did not include zero) were retained. The best fitting model included common A and unique AE moderation parameters only; common CE and unique C moderation parameters (and the residual C influence on externalizing disorders, i.e., c22) could be dropped without a decrement to model fit (fit statistics reported in row 3). The A moderation parameters could not be dropped without a decrement to model fit; thus, antisocial peer affiliation at age 17 moderated the additive genetic influences on externalizing disorders at age 17. Gene × Environment interaction results for the best fitting model are plotted in Figure 2a. The figure depicts how the additive genetic influence on externalizing disorders was greater in the context of greater antisocial peer affiliation. A similar pattern was also observed for nonshared environmental influences. Antisocial peer affiliation did not moderate the shared environmental influences on externalizing disorders.
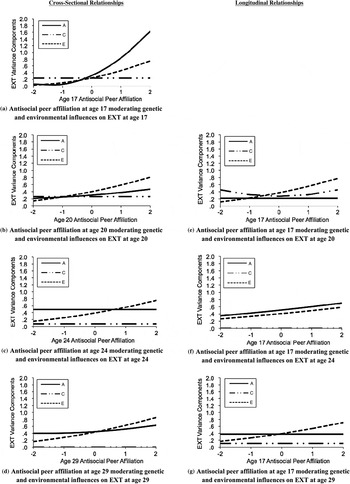
Figure 2. Antisocial peer affiliation moderating genetic and environmental influences on externalizing disorders: cross-sectional and longitudinal relationships. Changes in the unstandardized ACE variance components of externalizing disorders (EXT) are given as a function of antisocial peer affiliation for the best fitting models (see Table 5). A, additive genetic influence; C, shared environmental influences; E, nonshared environmental influence. The y-axis represents the unstandardized variance component score (shown for A, C, and E). The x-axis represents the value of antisocial peer affiliation (shown in 0, ±1, and ±2 SD). All composites were adjusted for age, sex, Age × Sex, and age2 by regressing these covariates out prior to analysis. Presented are the (a) results for the cross-sectional associations at age 17, (b) cross-sectional associations at age 20, (c) results for the cross-sectional association at age 24, (d) cross-sectional associations at age 29, (e) results for the longitudinal association between adolescent peer affiliation (age 17) and early emerging adult EXT (age 20), (f) interaction results for the longitudinal association between adolescent peer affiliation (age 17) and late emerging adult EXT (age 24), and (g) interaction results for the longitudinal association between adolescent peer affiliation (age 17) and late young adult EXT (age 29).
Table 5. Fit statistics for gene–environment interplay models of antisocial peer affiliation and EXT at ages 17, 20, 24, and 29
Note: –2LL, –2 log likelihood; Δχ2, chi-square change; AIC, Akaike information criterion; BIC, Bayesian information criterion; DIC deviance information criterion; A, additive genetic effects; C, shared environmental effects; E, nonshared environmental effects; EXT, externalizing disorders. The baseline model of comparison used in chi-square difference tests is the full ACE moderation model, which allows for ACE moderation on all common and unique parameters. The Δχ2 is the difference between the –2LL in the baseline model (full ACE moderation) compared to the other modes tested.
Gene × Environment interaction in young adulthood
As depicted by the fit statistics in Table 5 (rows 4–12) and the plot of variance components in Figure 2(b–d), there was little evidence that antisocial peer affiliation moderated genetic influences on externalizing disorders at ages 20 (Fig. 2b), 24 (Fig. 2c), or 29 (Fig. 2d). Although the best fitting models at ages 20 and 29 included parameters for the moderation of genetic influences on externalizing disorders, the changes in additive genetic influence at ages 20 and 29 were much smaller in magnitude in comparison to the additive genetic changes at age 17 (see the additive genetic moderation line in Fig. 2b and d in comparison to the additive genetic moderation line Fig. 2a). However, and like the age 17 results, nonshared environmental influences on externalizing disorders were greater in the context of greater antisocial peer affiliation at ages 20, 24, and 29, and the effects sizes were moderate in magnitude across time.
Long-term Gene × Environment interaction
Finally, we tested whether antisocial peer affiliation at age 17 moderated the genetic and environmental influences on externalizing disorders at ages 20, 24, and 29. The results for the best fitting models are depicted in Figure 2e–g (model fit statistics are reported in Table 5, rows 13–24). There was no evidence that adolescent antisocial peer affiliation moderated genetic influences on adult externalizing disorders at ages 20 or 29. However, at age 24, there was evidence of longitudinal common genetic moderation such that the genetic influences on externalizing disorders at age 24 were greater in the context of a greater affiliation with antisocial peers at age 17. Thus, in this sole case, there was evidence for longitudinal genetic moderation. Nevertheless, the effect size at age 24 was clearly not as substantial as the cross-sectional results at age 17 (compare Fig. 2a and f). Across longitudinal analyses, unique nonshared environmental moderation was significant, such that greater antisocial peer affiliation in adolescence was associated with greater nonshared environmental influences on externalizing disorders in adulthood. Thus, longitudinal results were generally consistent with cross-sectional associations at ages 17, 20, 24, and 29.
To control for prior antisocial peer affiliation and externalizing disorders, we also fit Gene × Environment interaction models between externalizing disorders and antisocial peer affiliation in adulthood after regressing out adolescent externalizing and antisocial peer affiliation on the target phenotypes at the adult ages (i.e., models were fit using residualized scores). The results for these analyses are provided in online-only supplementary Tables S.1–S.3 and Figure S.1. All results were consistent with those reported for the unadjusted phenotypes in that there was no evidence for additive genetic moderation of externalizing disorders past age 17.
Finally, to evaluate whether effects depended on substance misuse versus general antisocial behavior, we tested Gene × Environment interaction models for substance use disorder symptom counts (see Figure 3) and adult antisocial behavior symptom counts in separate models (see Figure 4; detailed fit statistics shown in Table 6). Following results for the externalizing composite, for both substance use disorders and adult antisocial behavior, results showed clear evidence for additive genetic moderation for the cross-sectional relationships at age 17, such that the genetic influence was greater in the context of a greater degree of antisocial peer affiliation. For adult antisocial behavior, however, there was no evidence for genetic moderation for the cross-sectional relationships at age 20, 24, and 29 or for the longitudinal relationships between antisocial peer affiliation at age 17 and adult antisocial behavior at ages 20, 24, and 29 (see Figure 4). Conversely, for substance use disorders, there was evidence of both cross-sectional and longitudinal genetic moderation at each time point. As shown in Figure 3, the genetic moderation results were generally strongest in effect size for the cross-sectional relationship at age 17 (see moderation line in [a]) in comparison to the cross-sectional relationships at ages 20, 24, and 29 (see b–d) or the longitudinal associations (see e–g).
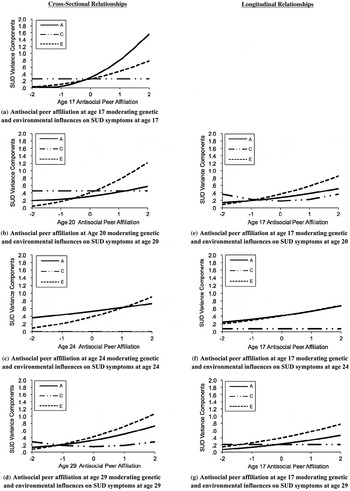
Figure 3. Antisocial peer affiliation moderating genetic and environmental influences substance use disorder (SUD) symptoms: Cross-sectional and longitudinal relationships. Changes in the unstandardized ACE variance components of SUD symptoms are given as a function of antisocial for the best fitting models (see Table 4). The y-axis represents the unstandardized variance component score (shown for A, C, and E). The x-axis represents the value of antisocial peer affiliation (shown in 0, ±1, and ±2 SD). A, additive genetic influence; C, shared environmental influences; E, nonshared environmental influence. All composites were adjusted for age, sex, Age × Sex, and age2 by regressing these covariates out prior to analysis. Presented are the (a) results for cross-sectional associations at age 17, (b) results for cross-sectional associations at age 20, (c) results for the cross-sectional association at age 24, (d) the cross-sectional associations at age 29, (e) results for the longitudinal association between adolescent peer affiliation (age 17) and early adult substance use disorder (age 20), (f) interaction results for the longitudinal association between adolescent peer affiliation (age 17) and late early adult substance use disorder (age 24), and (g) interaction results for the longitudinal association between adolescent peer affiliation (age 17) and young adult substance use disorder (age 29).
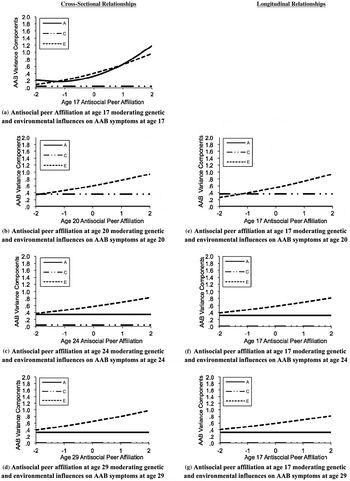
Figure 4. Antisocial peer affiliation moderating genetic and environmental influences on adult antisocial behavior symptoms (AABs): cross-sectional and longitudinal relationships. Changes in the unstandardized ACE variance components of AABs are given as a function of antisocial for the best fitting models (see Table 4). A, additive genetic influence; C, shared environmental influences; E, nonshared environmental influence. The y-axis represents the unstandardized variance component score (shown for A, C, and E). The x-axis represents the value of antisocial peer affiliation (shown in 0, ±1, and ±2 SD). All composites were adjusted for age, sex, Age × Sex, and age2 by regressing these covariates out prior to analysis. Presented are the (a) results for cross-sectional associations at age 17, (b) results for cross-sectional associations at age 20, (c) results for cross-sectional association at age 24, (d) cross-sectional associations at age 29, (e) results for the longitudinal association between adolescent peer affiliation (age 17) and early adult antisocial behavior symptoms (age 20), (f) interaction results for the longitudinal association between adolescent peer affiliation (age 17) and late early adult antisocial behavior symptoms (age 24), and (g) the interaction results for the longitudinal association between adolescent peer affiliation (age 17) and young adult antisocial behavior symptoms (age 29).
Table 6. Fit statistics for gene-environment interplay models of antisocial peer affiliation and externalizing disorders, shown separately for substance use disorder and adult antisocial behavior symptoms at ages 17, 20, 24, and 29
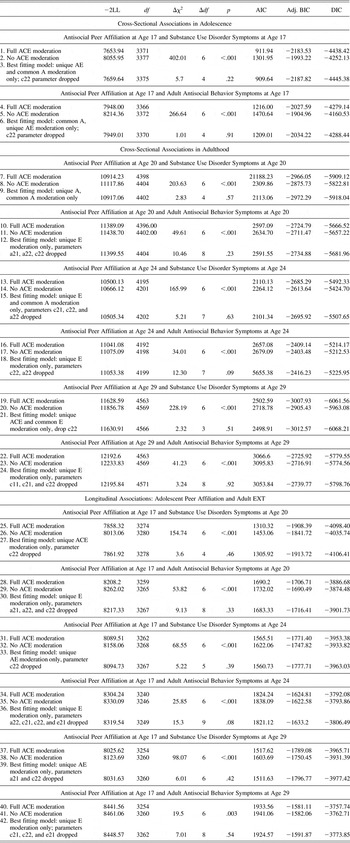
Note: –2LL, –2 log likelihood; Δχ2, chi-square change; AIC, Akaike information criterion; BIC, Bayesian information criterion; DIC deviance information criterion; A, additive genetic effects; C, shared environmental effects; E, nonshared environmental effects; EXT, externalizing disorders. The baseline model of comparison used in chi-square difference tests is the full ACE moderation model, which allows for ACE moderation on all common and unique parameters. The Δχ2 is the difference between the –2LL in the baseline model (full ACE moderation) compared to the other modes tested. In all best-fitting models, all parameters were significantly different from zero.
Discussion
Previous studies have shown evidence of both gene–environment correlation and interaction involving child and adolescent environmental contexts and adolescent externalizing problems (Benner et al., Reference Benner, Kretsch, Harden and Crosnoe2014; Byrd & Manuck, Reference Byrd and Manuck2014; Cicchetti et al., Reference Cicchetti, Rogosch and Thibodeau2012; Cleveland et al., Reference Cleveland, Wiebe and Rowe2005; Feinberg et al., Reference Feinberg, Button, Neiderhiser, Reiss and Hetherington2007; Hicks et al., Reference Hicks, South, Dirago, Iacono and McGue2009), but fewer studies have evaluated whether these processes are specific to adolescence or are also present in young adulthood. Here, we extended these findings by examining the consistency of Gene × Environment interplay involving antisocial peer affiliation and externalizing disorders across adolescence (age 17), early adulthood (ages 20 and 24), and later adulthood (age 29).
It is important to point out that overall genetic and environmental influences on externalizing disorders are generally comparable to earlier studies (Bergen, Gardner, & Kendler, Reference Bergen, Gardner and Kendler2007; Rhee & Waldman, Reference Rhee and Waldman2002). We showed that the heritability estimate of externalizing disorders was generally stable from ages 17 to 29 (range = ~45%–55%), although in that same age period, shared environmental influences decreased and nonshared environmental influences increased. A similar pattern was found for antisocial peer influences across time, although shared environmental influences remained significant and moderate in effect size through age 24. Genetic influences on antisocial peer affiliation were stable and moderate in magnitude from age 17 to 24 (estimates arranged from ~20% to 30%) and substantial by age 29 (51%). This is somewhat different than earlier studies (Kendler et al., Reference Kendler, Jacobson, Gardner, Gillespie, Aggen and Prescott2007), which reported substantial heritability estimates (~50%) from ages 12 to 25. Differences may be due to sample characteristics. For example, Kendler et al. used males only and retrospective report whereas we used equivalent numbers of males and females and prospective reports. Nonetheless, the results across both the Kendler et al. study and this study show nearly half the total variance in antisocial peer affiliation by age 29 is accounted for by additive genetic factors.
More important, and following previous research on parenting (Samek, Hicks, et al., Reference Samek, Hicks, Keyes, Bailey, McGue and Iacono2015) and school factors (Johnson et al., Reference Johnson, McGue and Iacono2009), we failed to detect any meaningful genetic moderation as a function of greater antisocial peer affiliation beyond age 17. This was true for our evaluation of the externalizing disorder composite and in subsequent evaluations of antisocial behavior alone. However, for symptoms of substance use disorders, we found evidence of genetic moderation as a function of greater antisocial peer affiliation from ages 17 through 29. The largest effect sizes for genetic moderation of substance use disorders were found at age 17, with comparatively smaller but nonetheless significant moderating genetic influences at ages 20, 24, and 29. Thus, results support the notion that antisocial peers continue to have a moderating influence on genetic and environmental risk for substance use disorders through young adulthood; perhaps because substance use disorders are more common in early and later adulthood relative to antisocial behavior (e.g., Moffitt, Reference Moffitt1993; Substance Abuse and Mental Health Administration, 2013), exposure to antisocial peers continues to have an important socializing effect.
These results have a number of important implications. The first is that, given the differences in findings by substance use disorder in comparison to adult antisocial behavior alone, it remains important for future research to evaluate individual facets of externalizing disorders in addition to a conglomerate externalizing measure. Our findings suggest this may be especially true for analyses involving externalizing disorders in adulthood. The second is that greater expression of genetic risk for externalizing disorders as a function of greater antisocial peer affiliation seems to be particularly important in late adolescence. This is consistent with a previous study by Kendler et al. (Reference Kendler, Gardner and Dick2011) that used retrospective reports of alcohol use and several environmental variables and detected a similar Gene × Environment interaction in early and middle adolescence, but not in adulthood. Here, we confirmed what can be described as a Gene × Environment × Development interaction effect between externalizing disorders (particularly the adult antisocial symptom criteria of adult antisocial personality disorder) and antisocial peer affiliation using a longitudinal design. This finding is consistent across other environmental variables; that is, other analyses using this sample have observed the same pattern for parent-relationship quality (Samek, Hicks, et al., Reference Samek, Hicks, Keyes, Bailey, McGue and Iacono2015), academic engagement (Johnson et al., Reference Johnson, McGue and Iacono2009), and prosocial peer affiliation (Samek, Hicks, Keyes, Iacono, & McGue, Reference Samek, Hicks, Keyes, Iacono and McGue2014). These findings suggest that adolescence may be a critical period for the emergence of externalizing disorders, when exposure to environmental risk factors seems to potentiate what may have been unexpressed genetic risk for externalizing psychopathology.
Following adolescence, gene–environment correlation or common genetic influences primarily account for the association between externalizing disorders and antisocial peer affiliation, as well as several other environmental risk factors (Johnson et al., Reference Johnson, McGue and Iacono2009; Samek, Hicks, et al., Reference Samek, Hicks, Keyes, Bailey, McGue and Iacono2015). That is, once genetic risk factors have been triggered by exposure to environmental risk, selection effects (i.e., active gene–environment correlation) appear to help maintain the mutual stability of externalizing disorders and high-risk environmental contexts. In addition, for substance use disorders but not adult antisocial behavior, we continued to detect a small Gene × Environment interaction of greater additive genetic and nonshared environmental variance as a function of greater antisocial peer affiliation. This suggests antisocial peers continue to have a potentiating influence on genetic and environmental risk for substance use disorders through young adulthood.
This study had several important limitations. One is that the sample had little racial and ethnic diversity. In addition, it is unclear if these results would replicate in more extreme samples (e.g., clinical or “at-risk” samples), although we might expect gene–environment interaction to be even more relevant to those experiencing extreme environmental adversity in adolescence, given our results on Gene × Environment interaction in adolescence. Another limitation is that the age ranges and environmental moderators we examined also limit the scope of our interpretations. For example, selection effects or gene–environment correlations and Gene × Environment interactions are likely to be present prior to age 17, and so it will be important to examine the stability of gene–environment interplay between externalizing disorders and contextual risk from childhood through adolescence. Relevant to this, prior research has shown that peer influences may be more critical in early or middle adolescence relative to later adolescence (Gardner & Steinberg, Reference Gardner and Steinberg2005; Steinberg & Monahan, Reference Steinberg and Monahan2007; Sumter et al., Reference Sumter, Bokhorst, Steinberg and Westenberg2009); thus, it could be Gene × Environment interaction is even more critical in earlier adolescence relative to later adolescence. This remains to be tested because we evaluated only one time point in late adolescence here.
Further, we relied on a self-report assessment of antisocial peers. Prior research has demonstrated differences in genetic and environmental influence on peer measures depending on the method employed to assess peer deviance. For example, Bullock et al. (Reference Bullock, Deater-Deckard and Leve2006) showed that coder impressions of deviant friendship processes tended to be influenced more by shared environmental rather than by heritable influences, while teacher-reported peer deviance tended to be influenced more by heritable influences than by shared environmental influences. It remains important for future research to explore whether findings may be impacted by other measurement methods, including direct observation. Finally, environmental variables other than peers (e.g., characteristics of romantic partners, marriage, and parenthood) may be important moderators of genetic risk for adult externalizing psychopathology, and this area remains important for future research to address. Strengths of the study include the large sample with equivalent numbers of males and females, prospective design and analysis, and inclusion of diagnostic measurement of externalizing symptoms.
In total, the results show that it is necessary to investigate gene–environment interplay broadly across development, because what may seem like a ubiquitous effect at one time point (e.g., adolescence) may operate differently at other developmental periods and across different (but highly correlated) traits. While the evidence is becoming more convincing, it remains necessary to evaluate other, possibly more salient adult environmental contexts as moderators of genetic risk of adult externalizing disorders (e.g., romantic partner relationship characteristics) to better understand the etiology of externalizing psychopathology across development.
Supplementary Material
To view the supplementary material for this article, please visit http://dx.doi.org/10.1017/S095457941600010.












