Child maltreatment represents a pathogenic relational environment that confers significant risk for maladaptation and psychopathology across both psychological and biological domains of development (Cicchetti & Lynch, Reference Cicchetti, Lynch, Cicchetti and Cohen1995; Cicchetti & Toth, Reference Cicchetti and Toth2015a, Reference Cicchetti and Toth2015b). The development sequelae accompanying child maltreatment not only result in adverse consequences during infancy, childhood, and adolescence, but also often initiate a negative developmental cascade that continues throughout the life span (Masten & Cicchetti, Reference Masten and Cicchetti2010). The proximal environment involving the nuclear family, as well as more distal factors associated with the community and culture more broadly, transact to undermine typical biological and psychological developmental processes in children who have experienced maltreatment (Cicchetti & Lynch, Reference Cicchetti and Lynch1993).
Child maltreatment ushers in motion a probabilistic path of epigenesis for abused and neglected children characterized by an increased likelihood of failure and disruption in the successful resolution of salient developmental tasks, resulting in a profile of relatively enduring vulnerability factors that increase the probability of the emergence of maladaptation and psychopathology (Cicchetti, Reference Cicchetti, Cicchetti and Carlson1989; Cicchetti & Lynch, Reference Cicchetti, Lynch, Cicchetti and Cohen1995; Vachon, Krueger, Rogosch, & Cicchetti, Reference Vachon, Krueger, Rogosch and Cicchetti2015). Long before psychopathological conditions appear in adulthood among maltreated individuals, a host of deviations in developmental processes are likely to have occurred during childhood.
Because maltreated children experience the extremes of caregiving casualty, they provide one of the clearest opportunities for scientists to discover the myriad ways in which psychological stressors can affect biological systems. Comparisons between maltreated and nonmaltreated children can elucidate our understanding of the caregiving processes that contribute to the development of regulated neurobiological systems. Numerous interconnected neurobiologic systems are negatively affected by the various stressors associated with maltreatment. Moreover, each of these neurobiological systems influences and is influenced by multiple domains of psychological and biological development. It is highly probable that a number of the symptoms and disorders that maltreated children develop eventuate in conjuction with disturbances and dysregulation in neurobiological systems.
Maltreated children are exposed to atypical caregiving and high stress. Two questions come to the fore. How does this affect gene regulation? What are the implications for mental and physical health? Epigenetics involves the investigation of changes in gene function that do not alter the DNA sequence, but instead provide an extra layer of transcriptional control that regulates gene activity and plays an important role in the acute regulation of genes in response to environmental changes. Epigenetics is a promising avenue for both basic and intervention research on child maltreatment (Szyf & Bick, Reference Szyf and Bick2013; Weder et al., Reference Weder, Zhang, Jensen, Yang, Simen and Jackowski2014). Epigenetic research thus has the potential to elucidate mechanisms to explain how maltreatment experiences confer risk for physical and mental health problems (Szyf & Bick, Reference Szyf and Bick2013; Toth, Gravener-Davis, Guild, & Cicchetti, Reference Toth, Gravener-Davis, Guild and Cicchetti2013; Yang et al., Reference Yang, Zhang, Ge, Weder, Douglas-Palumberi and Perepletchikova2013).
DNA is not a static entity as it was once thought to be (Mill, Reference Mill, Kendler, Jaffee and Romer2011; Szyf & Bick, Reference Szyf and Bick2013). Epigenetic processes play a dynamic role in regulating gene expression and are responsive to the environment (Mill, Reference Mill, Kendler, Jaffee and Romer2011; Roth, Reference Roth2013; Toth et al., Reference Toth, Gravener-Davis, Guild and Cicchetti2013). A number of epigenetic mechanisms have been proposed linking prenatal exposure to maternal stress and early infant outcomes (Monk, Spicer, & Champagne, Reference Monk, Spicer and Champagne2012; Roth, Reference Roth2013). In addition, postnatal environmental exposures, such as early adversity or the quality of mother and child interactions, also may lead to shifts in developmental trajectories via epigenetic pathways (Toth et al., Reference Toth, Gravener-Davis, Guild and Cicchetti2013).
The most investigated epigenetic mechanism in research on child maltreatment has been DNA methylation. Alterations in methylation, which manifest as changes in cytosine nucleotide–phosphate–guanine nucleotide (CpG) sites in the DNA sequence, may persist in a stable form over a long time. Alterations in the structure of chromatin influence gene expressions. Specifically, genes are switched off when chromatin is silent, and they are switched on (i.e., expressed) when chromatin is active. These dynamic states of chromatin are controlled by a number of processes, including epigenetic patterns of DNA methylation. Individual differences in DNA methylation thus are a potential biomarker of the contributions that environmental factors make to the divergence in phenotypes of maltreated individuals who possess similar genetic endowments.
Researchers examining methylation have adopted an epigenome-wide approach, as well as a focus on candidate genes of interest. Investigations that have utilized a genome-wide approach have found that these methylation changes occur at many gene loci (Szyf & Bick, Reference Szyf and Bick2013). The majority of human epigenetic investigations on child maltreatment have been conducted with adults and have assessed maltreatment retrospectively. For example, in a small sample of maltreated and nonmaltreated males (maltreated, n = 12; nonmaltreated, n = 28), Suderman et al. (Reference Suderman, Borghol, Pappas, Pereira, Pembrey and Hertzman2014) found genome-wide methylation profiles in adult DNA associated with child adversity that justify future exploration of epigenetic regulation as a mediating mechanism for long-term health outcomes.
Research with animals that experience abuse demonstrates that experience-induced changes in DNA methylation occur in brain circuits that have functional relevance to trajectories of normal and abnormal behavior. Accordingly, experience-driven DNA-methylation changes in maltreated individuals also have implications for disruptions in neural circuitry and behavior (Szyf & Bick, Reference Szyf and Bick2013). Likewise, using salivary DNA specimens, whole-genome methylation differences have been found between maltreated and nonmaltreated children at 2,868 CpG sites. A substantial number of genes implicated in prostate, colorectal, breast, colon, and ovarian cancer were contained in the set of genes that showed differential methylation between maltreated and nonmaltreated children (Yang et al., Reference Yang, Zhang, Ge, Weder, Douglas-Palumberi and Perepletchikova2013). The results of these investigations suggest that epigenetic mechanisms may be associated with risk for developing major health problems in later life (Yang et al., Reference Yang, Zhang, Ge, Weder, Douglas-Palumberi and Perepletchikova2013).
Several investigations have used a candidate gene approach to examine the effects of child abuse and neglect on methylation. Epigenetic regulation is a viable candidate mechanism through which caregiving behaviors, including child maltreatment, may exert long-term effects on hypothalamus–pituitary–adrenal activity and neuronal function (Szfy & Bick, Reference Szyf and Bick2013). Candidate genes utilized in this regard to date include the serotonin transporter gene, the glucocorticoid receptor gene (nuclear receptor subfamily 3, group C, member 1 [NR3C1]), and FK506 binding protein 5 gene (FKBP5). These studies have found that child maltreatment and traumatic experiences were associated with increased methylation of NR3C1 (Perroud et al., Reference Perroud, Paoloni-Giacobino, Prada, Olié, Salzmann and Nicastro2011; van der Knapp et al., Reference van der Knaap, Hudziak, Verbiest, Verhulst, Oldehinkel and van Oort2014; Yang et al., Reference Yang, Zhang, Ge, Weder, Douglas-Palumberi and Perepletchikova2013) and FKBP5. For example, McGowan et al. (Reference McGowan, Sasaki, D'Alessio, Dymov, Labonte and Szyf2009) found increased methylation of the exon 1F NR3C1 promotor among suicide victims with a history of child maltreatment compared to controls. Regarding FKBP5, Klengel et al. (Reference Klengel, Mehta, Anacker, Rex-Haffner, Pruessner and Pariante2013) found that FKBP5, an important functional regulator of the stress hormone system, increased the risk of developing stress-related psychiatric disorders in adulthood through allele-specific (i.e., a particular functional polymorphism), childhood trauma-dependent DNA methylation in functional glucocorticoid response elements of FKBP5. Additional research has begun to emerge regarding methylation in other genes, including dopaminergic genes such as dopamine receptor D2/ankyrin repeat and kinase domain containing 1 (DRD2/ANKK1) and child maltreatment. For instance, Groleau et al. (Reference Groleau, Joober, Israel, Zeramdini, DeGuzman and Steiger2014) found a trend-level increase in methylation of DRD2 among women with bulimia-spectrum disorders and a history of child sexual abuse compared to control women.
The present study examined genome-wide methylation in a large (N = 548) sample of maltreated and nonmaltreated children from low-socioeconomic stress backgrounds, as well as site-specific genes methylation. Parameters of maltreatment also were assessed (e.g., onset/developmental timing of maltreatment; see Barnett, Manly, & Cicchetti, Reference Barnett, Manly, Cicchetti, Cicchetti and Toth1993; Cicchetti & Barnett, Reference Cicchetti, Barnett, Grove and Cicchetti1991; Manly, Reference Manly2005).
Hypotheses
-
1. Maltreated children will evince significant differences in methylation across the epigenome, relative to demographically comparable nonmaltreated children.
-
2. Compared to nonmaltreated children, maltreated children will demonstrate greater adverse physical and mental health risk based on patterns of differential methylation associated with disorder outcomes.
-
3. Maltreated children will exhibit differential methylation of specific genes negatively associated with health outcomes, compared to nonmaltreated children.
-
4. Gender, ancestry, and genotype differences will be controlled or examined interactively with maltreatment status. Developmental timing of maltreatment experiences will be related to further variation in site-specific methylation.
Method
Participants
The participants in this investigation included 548 children (262 female, 286 male) who attended a research summer camp program designed for school-aged low-income maltreated (n = 298) and nonmaltreated (n = 250) children. Children were on average 9.40 years old (SD = 0.88). The sample was racially (67.7% Black, 20.6% White, and 11.7% Biracial or other race) and ethnically (20.6% were Latino) diverse. Informed consent was obtained from parents of maltreated and nonmaltreated children for the child's participation in the summer camp program and for examination of any Department of Human Services (DHS) records pertaining to the family.
Children in the maltreated group were recruited through a DHS liaison who examined Child Protective Services reports to identify children who had been maltreated and/or were part of a family with a history of maltreatment. Children living in foster care were not recruited for the current investigation because of the frequent instability in foster care placements. The DHS liaison contacted eligible families and explained the study. Parents who were interested in having their child participate provided signed permission for their contact information to be shared with project staff. These families were representative of those receiving services through the DHS. Comprehensive reviews of all DHS records for each family were conducted. Maltreatment information was coded by trained research staff and a clinical psychologist, using the Barnett et al. (Reference Barnett, Manly, Cicchetti, Cicchetti and Toth1993) nosological system for classifying child maltreatment. Coding is based on all available information and does not rely on DHS determinations.
Because maltreating families primarily have low socioeconomic status (National Incidence Study; Sedlak et al., Reference Sedlak, Mettenburg, Basena, Petta, McPherson and Greene2010), nonmaltreating families were recruited from those receiving Temporary Assistance to Needy Families in order to ensure socioeconomic comparability between maltreated and nonmaltreated families. A DHS liaison contacted eligible nonmaltreating families and described the project. Parents who were interested in participating signed a release allowing their contact information to be given to project staff for recruitment. The families were recruited as nonmaltreated families after comprehensive DHS record searches confirmed the absence of any documented child maltreatment. Families who received preventative DHS services due to concerns over risk for maltreatment were not included within the nonmaltreated comparison group. In order to further verify a lack of DHS involvement, trained research assistants interviewed the mothers of children recruited for the nonmaltreatment group using the Maternal Child Maltreatment Interview (Cicchetti, Toth, & Manly, Reference Cicchetti, Toth and Manly2003) and reviewed records in the year following camp participation to assure that all information had been assessed.
Children in the maltreated and nonmaltreated groups were comparable on a number of family characteristics (see Table 1). These include maternal education, χ2 (1, N = 545) = 2.91, p > .05, marital status, χ2 (3, N = 545) = 4.46, p > .05, and family history of receiving public assistance, χ2 (1, N = 544) = 0.81, p > .05.
Table 1. Family demographic characteristics

Note: All group contrasts were nonsignificant.
Procedures
Day camp procedures
Maltreated and nonmaltreated children were randomly assigned to groups of 10 same-sex and same-age peers. Within these groups 5 children were maltreated and 5 were nonmaltreated. Each group was led by three trained camp counselors who were unaware of child maltreatment status and study hypotheses. Children participated in recreational activities throughout the week. After child assent was obtained, children participated in research assessments conducted by trained research assistants. DNA samples via saliva also were obtained from children, as described below. All research assistants were unaware of child maltreatment status and study hypotheses.
Measures
Maltreatment Classification System (MCS)
The MCS (Barnett et al., Reference Barnett, Manly, Cicchetti, Cicchetti and Toth1993) is designed to assess individual children's maltreatment experiences. The MCS utilizes DHS records to make independent determinations of maltreatment. The MCS classifies the subtypes that each child experienced, frequency of occurrence, subtype severity, and developmental periods of occurrence in order to designate the recency, onset, and chronicity of maltreatment. Subtypes of maltreatment include neglect, emotional maltreatment, physical abuse, and sexual abuse. Neglect refers to failure to provide for the child's basic physical needs for adequate food, clothing, shelter, and medical treatment. Neglect also includes lack of supervision, moral–legal neglect, and educational neglect. Emotional maltreatment involves extreme thwarting of children's basic emotional needs for psychological safety and security. Examples include belittling and ridiculing the child, extreme negativity and hostility, child abandonment, suicidal or homicidal threats, and extreme negativity and hostility. Physical abuse involves nonaccidental physical injury to the child such as bruises, welts, burns, choking, and broken bones. Sexual abuse involves attempted or actual sexual contact between the child and caregiver for purposes of the caregiver's sexual satisfaction or financial benefit. Examples of sexual abuse range from exposure to pornography or adult sexual activity to sexual touching and fondling to forced intercourse with the child.
The MCS has demonstrated reliability and validity in classifying maltreatment in a number of studies (Bolger & Patterson, 2001; Dubowitz et al., Reference Dubowitz, Pitts, Litrownik, Cox, Runyan and Black2005; English et al., Reference English, Upadhyaya, Litrownik, Marshall, Runyan and Graham2005; Manly, Reference Manly2005; Smith & Thornberry, Reference Smith and Thornberry1995). DHS records were coded using the MCS by trained research staff and a clinical psychologist. All coders achieved adequate reliability before coding records used for the study. Kappas for the presence of each of the maltreatment subtypes ranged from 0.90 to 1.00; intraclass correlations for severity ratings of individual subtypes of maltreatment ranged from 0.83 to 1.0.
In the present study, 72.1% of the maltreated children had experienced neglect, 59.4% experienced emotional maltreatment, 27.2% physical abuse, and 8.7% experienced sexual abuse. Therefore, emotional maltreatment and neglect were pervasive throughout the sample while physical and sexual abuse occurred less frequently. Consistent with other samples of maltreatment, the majority of children in this study experienced more than one subtype of maltreatment. More specifically, 58.9% of children had experienced two or more subtypes of maltreatment (M = 1.75 SD = 0.72), and 10 out of the 15 possible combinations of the four maltreatment subtypes were present in the sample.
Developmental timing variables indicate the occurrence of maltreatment in discrete developmental periods, including infancy, toddlerhood, preschool, early school age, and later school age. This information is used to define developmental period of maltreatment onset and recency of maltreatment. We defined a categorization of onset and recency groups by dichotomizing onset into early (prior to age 5) and later (age 5 and older), and recency as recent (age 5 or older) and not recent (prior to age 5). These groupings then define three onset/recency groups used in analyses: early onset–not recent, early onset–recent, and late onset–recent.
DNA collection, extraction, and genotyping
Trained research assistants obtained DNA samples from participants by collecting saliva using the Oragene DNA Self-Collection kits. DNA was purified from 0.5 ml of Oragene-DNA solution using the DNA Genotek protocol for manual sample purification using prepIT-L2P. Sample concentrations were determined using the Quant-iT PicoGreen dsDNA Assay Kit (P7589, Invitrogen). Single nucleotide polymorphism (SNP) genotyping was conducted using Applied Biosystems TaqMan SNP Genotyping Assays for rs671 at Chr.12:112241766 in ALDH2 and (C__11703892_10) and rs1800497 at Chr.11:113270828 in ANKK1 (C___7486676_10). The NR3C1 gene mutation rs41423247, commonly known as Bcll, was genotyped using previously reported primer and probe sequences (1). Individual allele determinations were made using TaqMan Genotyping Master Mix (Applied Biosystems, Catalog 4371357) with amplification on an GeneAmp 9700 (Applied Biosystems) and analyzing the endpoint fluorescence using a Tecan M200 and data analyzed with JMP 8.0 (SAS, Inc.). Human DNA from cell lines was purchased from Coriell Cell Repositories for all representative genotypes and confirmed by sequencing using dye terminator cycle sequencing on an ABI 3130xl. These positive controls and no template controls were run alongside study samples representing 9% of the total data output. Any samples that were not able to be genotyped to a 95% or greater confidence were repeated under the same conditions.
The call rate for the ALDH2 SNP rs671 was 99.8%. The frequency distribution of the ALDH2 SNP was GG = 100%. Because of the lack of genotypic variation for ALDH2, this SNP was not included in supplemental analyses. The call rate for the ANKK1 SNP rs1800497 was 99.6%. The ANKK1 SNP distribution did not deviate from Hardy–Weinberg equilibrium, χ2 (1) = 0.45, ns. The frequency distribution of the ALDH2 SNP was as follows: G/G = 47.3%, A/G = 42.0%, and A/A = 10.6%. A/G and A/A genotypes were combined for analyses. The call rate for the NR3C1 SNP was 99.8%. The frequency distribution did not deviate from Hardy–Weinberg equilibrium, χ2 (1) = 0.02, ns, and was as follows: C/C = 56.2%, C/G = 37.2%, and G/G = 6.4%. C/G and G/G were combined for analyses.
To address potential population stratification, ancestral proportion testing was conducted. DNA from study participants was subjected to SNP genotyping of the Burchard et al. panel of 106 SNPs (Lai et al., Reference Lai, Tucker, Choudhry, Parnell, Mattei and García-Bailo2009; Yaeger et al., Reference Yaeger, Alvial-Bront, Abdul, Nolan, Grann and Birchette2008), known to be informative for ancestry from Africa, Europe, and Native America. The SNPs were genotyped using the iPLEX platform from Sequenom Bioscience, Inc., which uses the Sequenom MassArray. Samples are subjected to single base primer extension (SBE) with fluorophore labeled nucleotides from primers designed for SNPs of interest. The samples including the SBE products were placed on the iPLEX platform and matrix-assisted laser desorption/ionization time of flight was used to identify the allele based on the fluorophore passing the detector at the expected time associated with the mass of the SBE primer. The SNP genotyping results were then recoded and uploaded into STRUCTURE v2.3.4, which uses algorithms developed by Pritchard and colleagues (Falush, Stephens, & Pritchard, Reference Falush, Stephens and Pritchard2003, Reference Falush, Stephens and Pritchard2007; Hubisz, Falush, Stephens, & Pritchard, Reference Hubisz, Falush, Stephens and Pritchard2009). Three SNP tests were excluded based on high allele call rates of the non-DNA containing wells. The data from the remaining 103 loci were uploaded into the software and set to analyze with an Admixture model of ancestry and initialization of the simulation on the GALA cohort (initialize of POPINFO). The simulation was set to run with a burn-in of 10,000, 1,000 Markov chain Monte Carlo repetitions, and assuming three populations within the group. The results of the simulations were subsequently identified as percent association to each ancestry group based on the known ancestry of the GALA cohort.
To facilitate Maltreatment × Race interaction tests, a grouping variable using ancestral proportion continuous scores was created with multinomial logistic regression to classify cases. Parent-reported race/ethnicity (coded 1 = African American, 2 = Caucasian, 3 = Hispanic, and 4 = other race/ethnicity) was predicted from proportion African ancestry and proportion Native American ancestry. Given the large proportion of African American children in our sample, we then created a binary variable to classify those with predominately African ancestry (n = 361, 65.9%) versus other ancestral compositions (n = 187, 34.1%).
DNA methylation
Salivary DNA samples were collected from participants using Oragene DNA collection tubes (DNA Genotek®). DNA was later isolated from 450 μl of Oragene-DNA/saliva solution using the PrepIT-L2P protocol. The diluted DNA samples were submitted to the BioMedical Genomics Center (BMGC) at the University of Minnesota for quality analysis and testing of whole-genome methylation analysis using the HumanMethylation450 BeadChip (Illumina). The samples were assayed for quality by determining the concentration, using the Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen, Item P7589) and real-time polymerase chain reaction (TaqMan) quantification of human DNA concentration. All samples passed BMGC quality control standards, and a normalized 0.5 μg human DNA for each participant was utilized in the subsequent methylation analyses.
Each 0.5 μg DNA sample was subjected to bisulfite conversion using the EZ-96 DNA Methylation Kit (Zymo Research, D5003) that converts unmethylated cytosine bases to uracils. This method utilizes the methyl group attached to a cytosine as a protecting group to deamination and subsequent conversion to a uracil. After bisulfite conversion, the total amount of DNA was increased by methylation-specific amplification using a whole-genome amplification process, which copies the converted uracils to thymine bases. The DNA was then enzymatically fragmented in an end-point fragmentation process.
Microarray processing and analysis of the Illumina Infinium HumanMethylation 450K BeadChip was also done by the University of Minnesota's BMGC. This covers over 485,000 individual sites with single nucleotide resolution of CpG sites both inside and outside CpG islands. The 450K BeadChip offers comprehensive genome-wide coverage including 99% of RefSeq genes with high quality by using more than 600 negative controls. Bisulfite-converted samples were then hybridized to these BeadChips followed by washing and staining per protocols prescribed by Illumina. The microarray bead chips were then imaged using a HiScan SQ system.
The fluorescence data was subsequently analyzed using the Methylation Module v1.9.0 of the GenomeStudio software package v2011.1 (Illumina). All data was background corrected and negative control normalized, producing average beta values. This average beta value represents the relative quantity of methylation at an individual site ranging from 0 to 1 (unmethylated to completely methylated). Tests that produced different results from technical replicates, originating from the same source individual and collection type, of study participant samples were identified as poor and removed from subsequent analyses. This was accomplished by using differential methylation analysis of replicate sample-average beta. Criteria for exclusion of CpG loci based on lack of precision within technical replicates was identified by selecting sites with |DiffScore| > 13, which is equivalent to a p value of <.01. Tests corresponding to these suspect loci (N = 5,244), those tests with p values of greater than .01 (N = 1,603), and SNP tests (N = 65) were excluded (N = 6,638, 1.4%). Beta values were analyzed using principal component analysis in Partek Genomics Suite (Partek Inc.) software. Review of the data distribution identified two samples as outliers that were subsequently removed from further analyses.
Data analytic plan
Differential methylation analysis of beta values was performed comparing maltreated to nonmaltreated children after subtracting background noise and normalizing to array controls using GenomeStudio, Methylation Module, and the Illumina Custom Model. The resulting measure of this calculation set is delta beta, which represents the amount of change to the average beta at a site, or relative percentage methylation difference between defined groups. Positive delta beta values indicate an elevation in relative methylation and a negative value indicates a reduction. Because there are males and females in both the maltreated and the nonmaltreated groups, the X and Y chromosome data were removed from subsequent analyses. To correct for multiple testing, the significance threshold to determine differential methylation was set to 5.0 × 10–7, which is consistent with prior research (i.e., Yang et al., Reference Yang, Zhang, Ge, Weder, Douglas-Palumberi and Perepletchikova2013) and recommendations by Raykan, Down, Balding, and Peck (Reference Raykan, Down, Balding and Peck2011). To investigate the association of child maltreatment with known disease biomarkers, the delta beta values of differentially methylated loci (p < 5.0 × 10–7) were then analyzed using GeneGo MetaCore Software (Thomas-Reuters, MetaCore Version 6.23).
Site-specific methylation analyses were performed using SPSS v.21. Prior to conducting analyses, beta values were transformed using the M-value method, which has been shown to be more statistically valid for differential analyses of methylation levels compared to beta values (Du et al., 2010). Analyses of the three genes under investigation (i.e., ALDH2, ANKK1, and NR3C1) progressed as follows:
-
1. For ALDH2, because of multiple CpG sites in the first exon region, exploratory factor analyses (EFAs) with Promax rotation were conducted for data reduction purposes.
-
2. Independent t tests were conducted to compare methylation levels at specific CpG sites among maltreated and nonmaltreated children.
-
3. Analyses of covariance (ANCOVAs) were then conducted examining maltreatment effects on methylation levels (maltreatment as a binary yes/no variable) including child gender and race as covariates to determine if significant maltreatment differences in methylation levels were statistically significant with the inclusion of these relevant covariates and to determine whether sex and/or race may moderate maltreatment effects on methylation levels.
-
4. Finally, ANCOVAs were tested to determine the effect of maltreatment timing (onset and recency) on methylation levels, controlling for child age and sex and all interactions.
Maltreatment was operationalized in two ways depending on analyses. For the first set of ANCOVAs, maltreatment was treated as a binary variable (yes/no). For the second set of ANCOVAs, maltreatment was operationalized into four groups: nonmaltreated (n = 250), early onset–not recent (n = 127), early onset–recent (n = 84), late onset–recent (n = 74) for the examining of the impact of maltreatment timing on methylation level.
Results
Preliminary analyses
Maltreated (n = 298) and nonmaltreated children (n = 250) did not differ on age, t (546) = –0.84, ns, and families’ current receipt of public assistance, χ2 (1) = 0.81, ns. Maltreated children were more likely to be male, 57.7%; χ2 (1) = 8.00, p = .005, and of non-African ancestry, 43.8%; χ2 (1) = 7.95, p = .005, compared to nonmaltreated children (45.6% male, 32.0% non-African ancestry).
Differential methylation analysis
The delta beta information was binned to 10% increments based on the expected amount of percent methylation from the nonmaltreated group, or expected average beta. The results indicated a pattern of greater methylation among maltreated children at 0.0–0.69 methylation and decreased methylation among maltreated children at ≥0.70 methylation (see Figure 1 for graphical representation). Data were further classified as low methylation (0%–29%), medium methylation (30%–69%), and high methylation (70%–100%). Table 2 illustrates that maltreated children had greater methylation at low methylation sites (n = 197 sites) and medium methylation sites (n = 730 sites) and less methylation at high methylation sites (n = 907 sites). Finally, the mean difference in methylation between the maltreated and nonmaltreated participants was 6.2% with a range of 1%–64% for the differentially methylated loci (p < 5.0 × 10–7). The location within genes of significantly different CpG sites (p < 5.0 × 10–7) and associated select functional gene regions are reported in Table 3. This illustrates that much of the statistically significant CpG loci were found in intergenic and gene body regions (34.4% and 42.6% respectively). After controlling for multiple comparisons, maltreated and nonmaltreated children had significantly different methylation values at a total of 1,876 CpG sites (p < 5.0 × 10–7, all sites).

Figure 1. (Color online) Genome-wide methylation difference for maltreated children compared to expected methylation (nonmaltreated children).
Table 2. Genome-wide methylation difference for maltreated children compared to expected methylation (nonmaltreated children)

Table 3. Number of loci significantly different at p < 5.0 × 10−7 between the maltreated and nonmaltreated participants grouped by CpG location within gene and associated select functional gene regions

Note: CpG, Cytosine nucleotide–phosphate–guanine nucleotide; Diff., different; Differen., differentially; UTR, untranslated region.
Disease by biomarker analysis
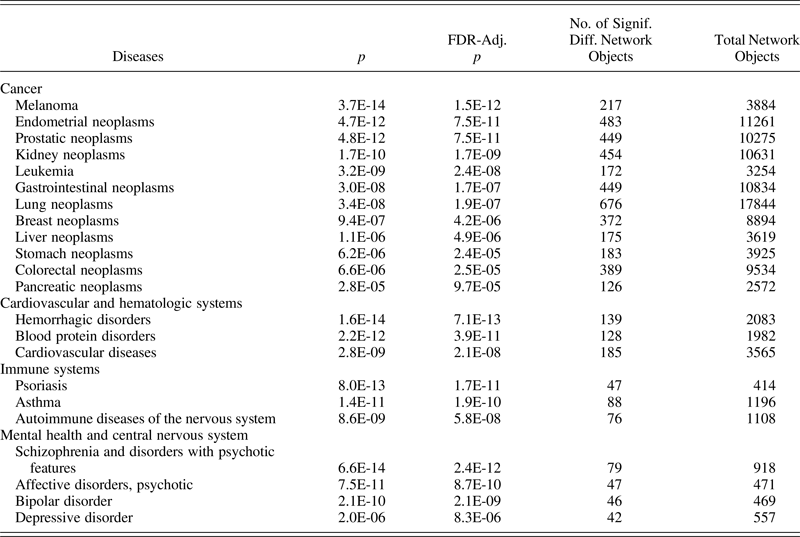
To investigate the potential association of maltreatment with known diseases, as indexed by genes, proteins, and other biomarkers, the delta beta values of differentially methylated loci (p < 5.0 × 10–7) from the HumanMethylation450 BeadChip were analyzed using GeneGo MetaCore Software (Thomas-Reuters, MetaCore Version 6.23). The GeneGo MetaCore software uses a Fisher exact test with Benjimini–Hochenberg false data recovery rate corrections. For each health index, Table 4 lists the number of network objects associated with the health index and the number of objects that were differentially methylated for maltreated versus nonmaltreated groups. The significance values for differential methylation rates are lower than the adjusted false discovery rate (p values, indicating significant maltreated/nonmaltreated group differences). A large number of network objects (i.e., proteins, peptides, and other biologically functional molecules) previously associated with mental health, cancer, cardiovascular systems, and immune functioning were identified evidencing differential methylation among maltreated and nonmaltreated children.
Table 4. Select disease indices that are differentially methylated among maltreated and nonmaltreated children

Note: FDR-Adj., False discovery rate adjusted; Diff., different.
Site-specific analyses
ALDH2
An EFA was conducted with CpG sites cg10449070, cg13955512, cg18780217, cg21470387, and cg24546205 from the first exon region. The results were not indicative of a one- or two-factor solution; therefore, each CpG site was analyzed separately. Independent samples t tests revealed a significant difference between maltreated and nonmaltreated children at site cg24546205, t (535) = –2.27, p = .02, such that maltreated children evidenced significantly higher methylation than nonmaltreated children. There were no significant differences between maltreated and nonmaltreated children on methylation level at the other four ALDH2 sites.
Next, full-factorial ANCOVAs including maltreatment (yes/no), gender, and race were conducted with the five ALDH2 CpG sites as outcomes in separate models. Even with the inclusion of gender, race, and all interactions, maltreated children continued to evidence significantly higher methylation at site cg24546205 compared to nonmaltreated children, F (1, 528) = 4.92, p = .03. No other significant maltreatment main effects were found for ALDH2 loci. Girls evidenced significantly higher methylation levels at site cg18780217, F (1, 538) = 4.44, p = .04, compared to boys. No other significant gender main effects were found for the ALDH2 CpG sites. There were no significant race main effects at any of the ALDH2 loci.
For cg13955512, a significant Maltreatment × Gender interaction was found, F (1, 536) = 15.41, p < .001 (see Figure 2a). Probing the interaction revealed that maltreated girls evidenced significantly lower methylation compared to nonmaltreated girls, F (1, 255) = 7.97, p = .005, and maltreated boys evidenced significantly higher methylation compared to nonmaltreated boys, F (1, 281) = 7.32, p = .007. Probed differently, results indicated that among nonmaltreated children, there was no difference in methylation level between boys and girls at this site. Among maltreated children, however, results indicated that boys evidenced significantly higher methylation compared to girls, F (1, 290) = 17.45, p < .001. None of the other interactive effects were significant in any models.

Figure 2. Effect of (a) maltreatment status and (b) developmental timing on ALDH2 methylation varied by gender. Mal, Maltreated; NMal, nonmaltreated; ENR, early onset–not recent; ER, early onset–recent; LR, late onset–recent. *p < .05. **p < .01.
To examine the role of maltreatment timing (onset and recency), the above ANCOVAs were repeated, substituting the binary maltreatment yes/no variable for the four-group maltreatment timing variable described above. None of the maltreatment timing main effects were significant for any of the ALDH2 loci. The results indicated a significant main effect of gender at cg13955512, F (1, 515) = 11.45, p < .001. The main effect of gender at cg13955512 was further clarified by a significant Maltreatment Timing × Gender interaction, F (3, 515) = 5.44, p < .001 (see Figure 2b). Probing the interaction revealed a significant effect of maltreatment timing among boys, F (3, 268) = 2.99, p = .03. Bonferroni contrasts indicated that nonmaltreated boys evidenced significantly lower methylation compared to early onset–not recently maltreated boys (p = .046). None of the other Bonferroni contrasts reached significance. Although there was also a significant effect of maltreatment timing among girls, F (3, 247) = 2.75, p = .04, none of the Bonferroni contrasts reached statistical significance. Probed differently, results indicated that among nonmaltreated children, there was no difference in methylation level between boys and girls. However, among children with early onset–not recent maltreatment, F (1, 120) = 7.59, p = .007, early onset–recent maltreatment, F (1, 80) = 4.62, p = .04, and late onset–recent maltreatment, F (1, 69) = 6.99, p = .01, boys consistently demonstrated significantly higher methylation compared to girls. There were no significant main effects of race, and none of the other interactions were significant for any of the other ALDH2 CpG sites.Footnote 1
ANNK1
Because there was only one CpG site in the first exon region for ANKK1, an EFA was not conducted. The results of an independent samples t test indicated that maltreated children evidenced significantly higher methylation than nonmaltreated children at site cg06976250, t (546) = –2.08, p = .04. An ANCOVA examining the effect of maltreatment status controlling for race, gender, and all interactions continued to support a significant difference between nonmaltreated and nonmaltreated children at this CpG site, F (1, 539) = 9.04, p = .003. Moreover, girls evidenced significantly higher methylation than boys, F (1, 539) = 9.26, p = .002, and children with African ancestry evidenced significantly higher methylation than children of non-African ancestry, F (1, 539) = 13.08, p < .001. None of the interactions were statistically significant.
To investigate the role of maltreatment timing (onset and recency) on methylation at ANKK1, the above ANCOVA was repeated substituting the binary maltreatment yes/no variable for the four-group maltreatment timing variable. A significant effect of maltreatment timing was found, F (3, 518) = 3.70, p = .01 (see Figure 3). Bonferroni contrasts indicated that children with early onset–non recent maltreatment evidenced significantly higher methylation compared to nonmaltreated children (p = .02). None of the other Bonferroni contrasts were statistically significant. As found in the previous model, girls evidenced significantly higher methylation compared to boys, F (1, 518) = 7.31, p = .007, and children with African ancestry evidenced higher methylation compared to children of non-African ancestry, F (1, 518) = 8.16, p = .004.Footnote 2

Figure 3. The effect of maltreatment developmental timing on ANKK1 methylation. Significant contrast: early onset–not recent > nonmaltreated. p < .05.
NR3C1
An EFA was conducted with CpG sites cg20753294, cg18146873, cg08818984, and cg26720913 from the first exon region for NR3C1. The results were not indicative of a one or two-factor solution; therefore, each CpG site was analyzed separately. Independent samples t tests did not support significant differences at any NR3C1 site between maltreated and nonmaltreated children. Furthermore, ANCOVAs including gender and race also did not indicate any significant main effects of maltreatment status. Girls evidenced significantly higher methylation at cg08818984, F (1, 539) = 5.67, p = .02, and cg26720913, F (1, 538) = 6.36, p = .01, compared to boys. None of the other main effects of maltreatment status, gender, or race were statistically significant. For site cg18146873, there was a significant maltreatment by race interaction, F (1, 539) = 4.50, p = .03 (see Figure 4). Probing the interaction revealed that maltreated children with African ancestry evidenced significantly lower methylation compared to nonmaltreated children with African ancestry, F (1, 333) = 5.65, p = .02. The effect of maltreatment status was nonsignificant for children of non-African ancestry, F (1, 206) = 0.72, ns. Probed differently, the results indicated that among nonmaltreated children, there was no difference between children of African ancestry and other children, F (1, 246) = 0.66, ns. However, among maltreated children, those of African ancestry evidenced significantly lower methylation compared to maltreated children of other ancestral backgrounds, F (1, 293) = 5.46, p = .02. None of the other interactive effects were significant at any of the NR3C1 sites.

Figure 4. The effect of maltreatment status on NR3C1 methylation varied by race. Mal, maltreated; NMal, nonmaltreated. *p < .05.
The next set of ANCOVAs examined the effect of maltreatment timing on methylation controlling for gender and race and all interactions. No significant main effects of maltreatment timing or race were found for any NR3C1 sites. Boys evidenced significantly lower methylation at sites cg08818984, F (1, 518) = 4.30, p = .04, and cg26720913, F (1, 517) = 5.14, p = .02, compared to girls. For site cg18146873, there was a significant maltreatment timing by gender interaction, F (3, 518) = 3.46, p = .02. However, probing the interaction revealed no significant contrasts. There were no other significant interactions for any NR3C1 CpG sites.Footnote 3
Discussion
This investigation was conducted on one of the largest samples to date of methylation differences between maltreated and nonmaltreated children. Because most of the studies of DNA methylation have been on adults who retrospectively reported having been maltreated, the fact that the participants in this investigation were maltreated children examined prospectively, and not adults, is very concerning. The sample was carefully recruited, with demographically comparable low-income children who have confronted the stress associated with poverty and adversity. The maltreated children all had Child Protective Services documented maltreatment experiences (Barnett et al., Reference Barnett, Manly, Cicchetti, Cicchetti and Toth1993; Cicchetti & Barnett, Reference Cicchetti, Barnett, Grove and Cicchetti1991; Manly Reference Manly2005), and we were able to ascertain the developmental timing of maltreatment events since the birth of the targeted child.
We found, as have Yang et al. (Reference Yang, Zhang, Ge, Weder, Douglas-Palumberi and Perepletchikova2013) and others, that there were significant differences between maltreated and nonmaltreated children in methylation across the epigenome. Maltreated children tended to have higher levels of methylation at CpG sites where nonmaltreated evinced low and medium levels of methylation, as well as lower methylation levels at CpG sites where nonmaltreated children evinced high levels of methylation (see Table 2 and Figure 1). The pattern of methylation differences was related to differential physical and mental health risk for maltreated and nonmaltreated children. Maltreated children exhibited differential methylation in genes associated with various cancers, cardiovascular and hematologic disease, immune disorders, as well as major psychiatric disorders (e.g., schizophrenia, bipolar disorder, and depression). Thus, the genome-wide epigenetic study of child maltreatment demonstrates an increased risk for mental and physical disorders and suggests liabilities for future health outcomes.
In addition, we conducted site-specific analyses of methylation on CpG sites of three genes: ALDH2, ANKK1 (associated sites of DRD2), and NR3C1 to examine variation among maltreated children on these genes related psychological functioning and disorders. Not all maltreated children exhibit the same degree of methylation. The effects of developmental timing, gender, and race were evaluated. Analyses also controlled for genotype variation. The site-specific results were not altered by controlling for genotypic variation of respective genes.
For NR3C1 (the glucocorticoid receptor gene), among African American children, maltreated children evinced much lower methylation than nonmaltreated children, and among maltreated children, African American children had lower methylation than children who were not African American. Although prior studies have shown that early maltreatment and trauma are associated with increased methylation of NR3C1 (Perroud et al., Reference Perroud, Paoloni-Giacobino, Prada, Olié, Salzmann and Nicastro2011; van der Knapp et al., Reference van der Knaap, Hudziak, Verbiest, Verhulst, Oldehinkel and van Oort2014), the role of ancestral variation has not been well studied. Our findings suggest that this is an important consideration for future research.
The ANKK1 gene, also referred to as Taq1A, was originally associated with the DRD2 gene. Because of its close proximity to DRD2, ANKK1 is believed to regulate DRD2 (Samochowiec, Samochowiec, Puls, Bienkowski, & Schott, Reference Samochowiec, Samochowiec, Puls, Bienkowski and Schott2014). Our results suggest that maltreated children (boys and girls) with early onset but not recent maltreatment had significantly higher ANKK1 methylation than nonmaltreated children suggesting a potential dopaminergic pathway of maltreatment risk that may be related to the early experience of maltreatment. Similarly, given the documented association of ALDH2 and risk for alcohol use disorders (e.g., Dick & Foroud, Reference Dick and Foroud2003), our findings that maltreated boys with early onset but not recent maltreatment showed the largest methylation differences compared to nonmaltreated boys indicates another potential pathway of epigenetic risk.
Together, our site-specific findings add to the existing literature by demonstrating that the effects of child maltreatment on methylation in the glucocorticoid receptor gene (NR3C1), dopaminergic gene (ANKK1), and alcohol-metabolizing gene (ALDH2) are complex and highlight the need for examining individual characteristics such as gender, race, and developmental timing of maltreatment. Given the associations of these three genes with various psychological outcomes, explicating the role of epigenetic modifications of these genes in the development of psychopathology for maltreated children may be of critical importance.
Future directions
In the future, it will be important to study other manifestations of stress-related risk in maltreated children, such as allostatic load and constituent biomarkers and immune functioning, in order to understand how methylation may affect these processes related to future disease. Future epigenetic research needs to determine whether African American children are more protected by lower methylation of NR3C1 or if lower methylation of this gene suggests greater risk for mental and physical problems. In addition, research needs to examine linkages of differential methylation in maltreated children to behavioral outcomes. Maltreated children exhibit increased behavioral problems and psychopathology (Cicchetti & Toth, Reference Cicchetti, Toth and Cicchetti2016; Cicchetti & Valentino, Reference Cicchetti, Valentino, Cicchetti and Cohen2006; Vachon et al., Reference Vachon, Krueger, Rogosch and Cicchetti2015); methylation may serve as a mediator between maltreatment and adverse outcomes. Finally, investigations of other high-risk groups of children who experience varying degrees of inconsistent caregiving (e.g., Esposito et al., Reference Esposito, Jones, Doom, MacIssac, Gunnar and Kobor2016) will not only offer insight into the specificity of these neurobiological system dysregulations in response to maltreatment and its associated stressors but also contribute to the development of more knowledge of which aspects of caregiving experiences are critical for normal neurobiological growth (Toth, Sturge-Apple, Rogosch, & Cicchetti, Reference Toth, Sturge-Apple, Rogosch and Cicchetti2015).