


Interest in behavioral epigenetics is currently booming in response to its potential to identify mechanisms through which environmental exposures become biologically embedded to affect infant behavioral and psychological traits (Boyce & Kobor, Reference Boyce and Kobor2015; Kinsella & Monk, Reference Kinsella and Monk2009). This opportunity is afforded by recent breakthroughs in our understanding of, and ability to investigate, epigenetics (molecular processes occurring on and around the genome that regulate gene activity without changing the underlying DNA sequence; Bird, Reference Bird2007). Human developmental researchers studying epigenetic processes have already achieved notable successes linking maternal experience to prenatal origins of health, with robust case studies showing epigenetic mechanisms partially mediate the effects of some prenatal maternal exposures (e.g., nutrition) on subsequent child health and behavior (Heijmans et al., Reference Heijmans, Tobi, Stein, Putter, Blauw, Susser and Lumey2008; Painter et al., Reference Painter, Westendorp, de Rooij, Osmond, Barker and Roseboom2008; Tobi et al., Reference Tobi, Lumey, Talens, Kremer, Putter, Stein and Heijmans2009). Despite such advances, mechanisms underpinning associations between prenatal maternal mood disorder and newborn behavioral outcomes remain poorly understood. This is unfortunate given evidence that mood disorders are disproportionately prevalent among pregnant women and appear to confer risk for altered fetal development and early childhood behavior (Kinsella & Monk, Reference Kinsella and Monk2009).
We first provide a general overview of mechanisms by which prenatal exposure to maternal mood disorder may affect fetal and newborn neurobehavior in part via epigenetic processes. We then outline the two dominant approaches for examining epigenetic effects: the candidate epigenetic approach and epigenome-wide association studies (EWAS). We describe advantages and disadvantages of these approaches and briefly outline studies that have used these methods to examine associations between prenatal exposures and fetal and newborn behavioral outcomes. Recently, concerns have been raised about the use of the word epigenetics as being “ambiguous, over-encompassing, and uncoupled from its historical roots” (Greally, Reference Greally2018, p. 207). We respond to this important criticism by advancing a functional epigenetic pathway approach that capitalizes on the strengths, and minimizes the weaknesses, of both candidate epigenetic and EWAS methods.
Prenatal Exposure to Maternal Mood Disorder and Neurobehavioral Outcomes: The Role of the Hypothalamic–Pituitary–Adrenal (HPA) Axis
There is no question that exposure to maternal mood disorder during early childhood is a risk factor for the development of psychopathology in the child (Goodman et al., Reference Goodman, Rouse, Connell, Broth, Hall and Heyward2011). Exposure to maternal mood disorder, such as depression, is associated with altered neuroendocrine stress responses in offspring (Essex, Klein, Cho, & Kalin, Reference Essex, Klein, Cho and Kalin2002) and a more fearful and reactive temperament (Davis et al., Reference Davis, Glynn, Schetter, Hobel, Chicz-Demet and Sandman2007). These children are also more likely to exhibit internalizing symptoms in early childhood and anxiety in adolescence (Goodman et al., Reference Goodman, Rouse, Connell, Broth, Hall and Heyward2011). There is evidence that this risk may be partially transmitted from mother to child in utero (Monk et al., Reference Monk, Feng, Lee, Krupska, Champagne and Tycko2016). This route of risk transmission is especially important to understand given that fetal exposure to maternal mood disorder may program fetal and newborn neurobehavior and thus vulnerability to the development of psychological disorders later in life, a third pathway beyond shared genes and postnatal environmental exposures by which the next generation inherits familial vulnerability to psychopathology (Monk, Spicer, & Champagne, Reference Monk, Spicer and Champagne2012).
The HPA axis is the most widely studied mechanism linking prenatal exposure to maternal mood and newborn neurobehavior. These associations are far from clear, and there are additional mechanisms of interest, such as immune system functioning and the microbiome (Beijers, Buitelaar, & de Weerth, Reference Beijers, Buitelaar and de Weerth2014), but few have generated as much empirical evidence as the HPA axis. Pregnant women with mood disorders are more likely to have greater circulating levels of cortisol in the blood compared to their nondepressed peers (Murphy et al., Reference Murphy, Braithwaite, Hubbard, Williams, Tindall, Holmes and Ramchandani2015). They also exhibit greater concentrations of corticotropin releasing hormone (CRH) in cerebrospinal fluid, which are reduced following electroconvulsive therapy treatment (Kasckow, Baker, & Geracioti, Reference Kasckow, Baker and Geracioti2001). When the HPA axis is activated, CRH is released from the hypothalamus, which stimulates the synthesis of ACTH by the pituitary and leads to the eventual release of glucocorticoids (cortisol) by the adrenal glands (see Figure 1). However, there is substantial variability in mood disorders. More recent hypotheses suggest that pregnant women exhibiting “atypical” depression, characterized by rejection sensitivity, agitation, irritability, and mood reactivity, exhibit a blunted diurnal cortisol response and lower cortisol levels upon awakening (Glynn, Davis, & Sandman, Reference Glynn, Davis and Sandman2013).
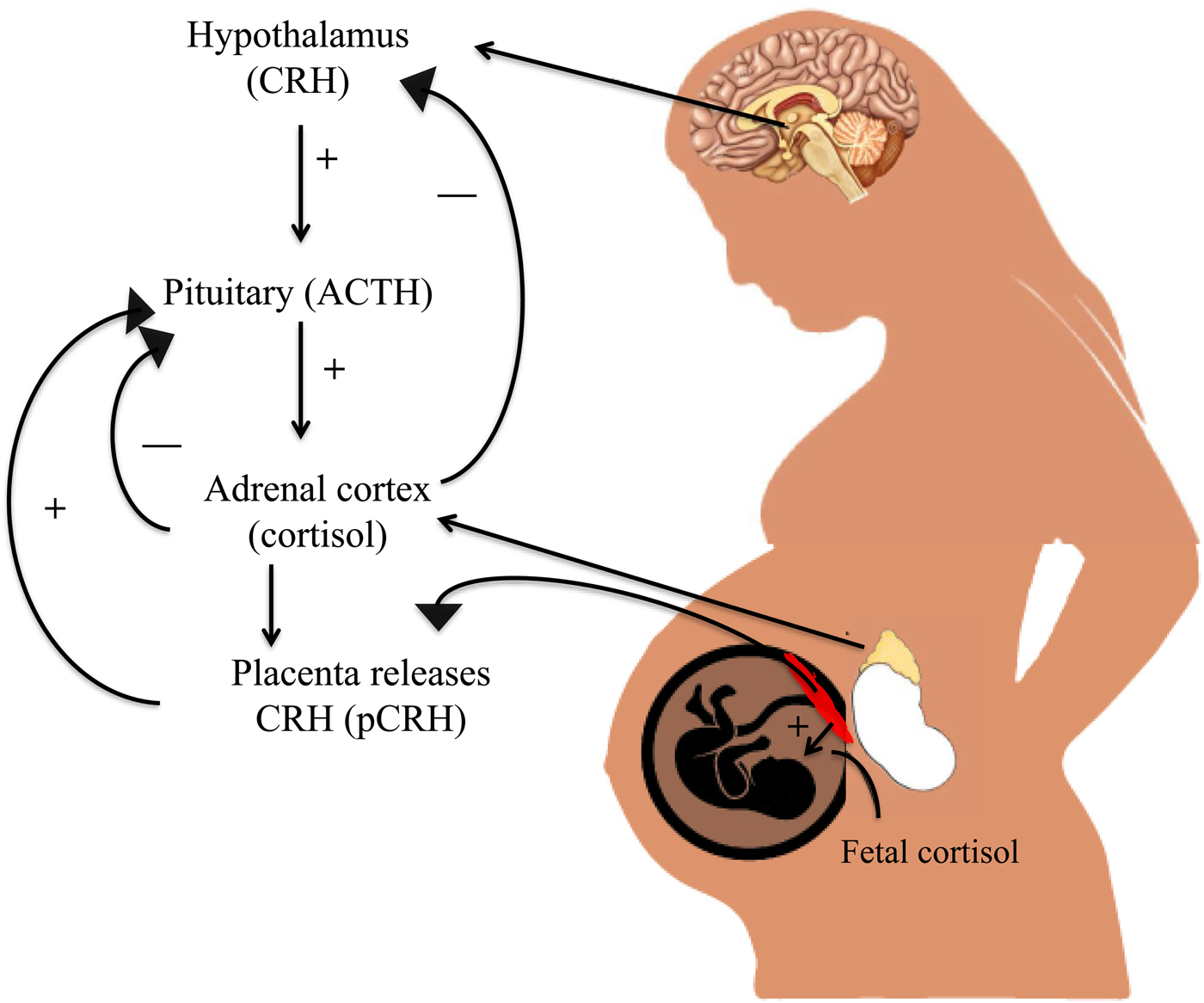
Figure 1. (Color online) Hypothalamic–pituitary–adrenal (HPA) axis regulation in the maternal–placental–fetal unit. During pregnancy, the placenta releases corticotropin-releasing hormone (CRH) into the fetal and maternal bloodstream. Maternal cortisol increases production of placental CRH, which rises across the course of gestation and leads to increases in maternal and fetal cortisol.
In typical human pregnancies, maternal cortisol levels are 5 to 10 times higher than cortisol levels of the fetus (Ishimoto & Jaffe, Reference Ishimoto and Jaffe2011). Higher than normal levels of circulating cortisol in the maternal bloodstream could cross the placenta (which typically filters out 80%–90% of maternal cortisol), thus exposing the fetus to excess cortisol and potentially reprogramming the fetal HPA axis (Sandman & Davis, Reference Sandman and Davis2012). When a pregnant woman experiences stress, glucocorticoids also activate CRH expression in the placenta, leading to greater synthesis and release of cortisol in the fetal and maternal bloodstream. The release of placental CRH does not inhibit the maternal HPA axis but instead leads to greater increases in cortisol until parturition (Figure 1; Sandman, Glynn, & Davis, Reference Sandman, Glynn, Davis, Reissland and Kisilevsky2016). There is a great deal of evidence suggesting that placental CRH production may be accelerated by an adverse intrauterine environment characterized by high levels of prenatal stress exposure (Sandman et al., Reference Sandman, Glynn, Davis, Reissland and Kisilevsky2016). Greater CRH release, for instance, occurs when placental cells are exposed to cortisol, catecholamines, and proinflammatory cytokines (Sandman & Davis, Reference Sandman and Davis2012), many of which are also associated with maternal mood disorder. These elevations could alter fetal brain structures such as the amygdala, as is the case in animal models (Salm et al., Reference Salm, Pavelko, Krouse, Webster, Kraszpulski and Birkle2004) and human studies (Buss et al., Reference Buss, Davis, Shahbaba, Pruessner, Head and Sandman2012).
CRH exposure also affects fetal HPA axis development, especially near term, by stimulating the release of fetal cortisol from the fetal adrenal axis (Sirianni, Rehman, Carr, Parker, & Rainey, Reference Sirianni, Rehman, Carr, Parker and Rainey2005). An increase in CRH, which accompanies typical human pregnancy, also impacts fetal responses to novelty and greater fetal heart rate reactivity, suggesting that the fetal nervous system is adversely affected by greater than typical increases in CRH (Sandman et al., Reference Sandman, Glynn, Davis, Reissland and Kisilevsky2016). There are also effects of maternal stress on newborn stress regulation, fearful temperament, and brain development (Sandman et al., Reference Sandman, Glynn, Davis, Reissland and Kisilevsky2016). Children exposed to high levels of maternal cortisol in utero were twice as likely to exhibit borderline or clinically significant levels of anxiety in childhood (Sandman & Davis, Reference Sandman and Davis2012) and had a right amygdala that was 6.4% larger than those with lower exposure (Buss et al., Reference Buss, Davis, Shahbaba, Pruessner, Head and Sandman2012). Greater exposure to high levels of maternal cortisol in the second half of gestation was also related to a larger, prolonged cortisol response to stress in neonates (Buss et al., Reference Buss, Davis, Shahbaba, Pruessner, Head and Sandman2012).
There are a number of important epigenetic mechanisms that may underlie the association of prenatal exposure to maternal mood disorder on fetal and newborn neurobehavioral outcomes, including histone modification, genomic imprinting, and DNA methylation. Here we focus on DNA methylation given recent breakthroughs using this platform in behavioral epigenetics (Weaver et al., Reference Weaver, Cervoni, Champagne, D'Alessio, Sharma, Seckl and Meaney2004), and the enhanced feasibility of integrating methylation assays into behavioral and biomedical studies (Adkins, Rasmussen, & Docherty, Reference Adkins, Rasmussen, Docherty and Hopcroft2017). DNA methylation refers to the attachment of a small molecule (i.e., methyl group) to a specific DNA position (i.e., genomic locus). While the full range of DNA methylation's functional effects are unknown, one of its primary functions is to silence the expression of the associated gene by blocking the interaction of regulatory molecules to the methylated DNA (Bird, Reference Bird2007). That is, when a methyl group is attached to a specific locus in a given gene's promoter region, specifically transcription factor binding sites, it can block the process of gene expression, therefore silencing the anticipated activity of that gene (Bird, Reference Bird2007). Conversely, methylation in other regions of the genome, gene bodies, for instance, can instead increase gene expression (Jones, Reference Jones2012).
Recent calls have been made to expand focus beyond the HPA axis (Beijers et al., Reference Beijers, Buitelaar and de Weerth2014), with which we concur. We therefore describe the functional epigenetic pathway targeting the HPA axis as a proof of principle, but describe how this pathway approach can generalize to any known pathway, or even examine the full range of known biological pathways (Docherty, Moscati, Adkins, et al., Reference Docherty, Moscati, Adkins, Wallace, Kumar, Riley and Bacanu2017; Docherty, Moscati, Dick, et al., Reference Docherty, Moscati, Dick, Savage, Salvatore, Cooke and Kendler2017). We argue that a bioinformatics approach to analyzing epigenetic data that focuses on a specific, theory-driven biological pathway of interest will advance the field of behavioral epigenetics and provide much needed mechanistic insights into how epigenetic processes could be implicated in the development of neurobehavioral disorders. We further highlight the importance of targeting CpG sites associated with known transcription factor binding sites, given that it is in these regions that DNA methylation is likely to lead to differences in gene expression (Bird, Reference Bird2007).
Below we review advantages and disadvantages of candidate epigenetic approaches, some of which have found associations between DNA methylation at transcription factor binding sites and newborn neurobehavior. We assert that this epigenetic pathway approach targeting transcription factor binding sites is likely to significantly advance our mechanistic understanding of biological embedding, and add much-needed specificity to programming theories related to prenatal exposures and neurobehavioral development (Greally, Reference Greally2018; Lappalainen & Greally, Reference Lappalainen and Greally2017).
Candidate Epigenetic Studies: Advantages and Disadvantages
Candidate epigenetic studies have, until recently, dominated the nascent field of behavioral epigenetics (Conradt, Reference Conradt2017; Lester, Conradt, & Marsit, Reference Lester, Conradt and Marsit2016). Much of the empirical work to date has focused on a single candidate: the glucocorticoid receptor gene, NR3c1, which is involved in reuptake of cortisol throughout the body, but most notably in the hippocampus (Herman & Spencer, Reference Herman and Spencer1998). Research into the topic began with the observation of maternal effects on offspring differences in behavior and HPA stress responses in rodent models (Meaney, Reference Meaney2010). In these models, maternal behavior was shown to influence HPA responses to stress in offspring through tissue-specific effects on gene expression (Weaver et al., Reference Weaver, Cervoni, Champagne, D'Alessio, Sharma, Seckl and Meaney2004), including glucocorticoid receptor expression in hippocampal brain areas. This region is of interest given that glucocorticoid activation inhibits HPA activity via negative feedback inhibition (Herman & Spencer, Reference Herman and Spencer1998). These findings were further supported by evidence that familial dysfunction and childhood adversity are also linked to altered HPA stress response in humans, which, in turn, is associated with increased risk for various types of psychopathology (De Bellis et al., Reference De Bellis, Chrousos, Dorn, Burke, Helmers, Kling and Putnam1994; Heim & Nemeroff, Reference Heim and Nemeroff2001). Building on this body of research, Meaney (Reference Meaney2010) combined rodent models with emerging epigenetic technologies to show that adult offspring who were born to mothers who engaged in low rates of maternal care show increased methylation at a glucocorticoid receptor gene (NR3c1) promoter in hippocampus, which upregulates HPA stress response in adulthood. Recently, increased methylation at the NR3c1 promoter has been observed in human suicide victims, and studied in relation to psychosocial stress in humans, with mixed results (McGowan et al., Reference McGowan, Sasaki, D'Alessio, Dymov, Labonté, Szyf and Meaney2009; Turecki & Meaney, Reference Turecki and Meaney2016).
It is noteworthy that the trauma–mood disorder link appears to be mediated partially by epigenetic modifications to NR3c1 in the hippocampus. This finding has served as an important proof of principle for behavioral epigenetic research, and consequently, attracted tremendous resources to epigenetic studies of candidate genes with known biological function. Thus, while this approach has yielded profound insights into the biological embedding of social adversity (Essex et al., Reference Essex, Thomas Boyce, Hertzman, Lam, Armstrong, Neumann and Kobor2013; Monk et al., Reference Monk, Spicer and Champagne2012), its success has also led to an overly narrow focus on candidate epigenetic studies, particularly those related to glucocorticoid function. This emphasis on candidate epigenetic studies was due, in part, to the once prohibitive cost of EWAS; however, recent technological advances have rendered these studies cost-effective and easily implemented (Adkins et al., Reference Adkins, Rasmussen, Docherty and Hopcroft2017).
Nonetheless, a major advantage of the candidate epigenetic approach is that these studies are theoretically driven with clear hypothesized relations between gene function and outcome. They also provide more precise mechanistic insights into the nature of how environmental exposures or insults may affect later child behavior (Keating, Reference Keating2016).
The principal disadvantage of candidate epigenetic studies is that they likely oversimplify what is known to be an extremely complex process. While once necessitated by technological limits, it has long been recognized that focus on a single gene, locus, or epigenetic mark suffers a profound research limitation in its disregard for the well-established, highly multifactorial, polygenic etiology of complex traits (Lester et al., Reference Lester, Conradt and Marsit2016). In addition, maternal mood disorder is complex and can be associated with multiple comorbid diagnoses (e.g., personality disorders), health practices (e.g., prenatal care), and risk factors (e.g., secondhand smoke exposure). To date there has been a seemingly arbitrary focus on select CpG sites (or a range of CpG sites) within a candidate gene, with relatively little theoretical justification for the selection of those sites, which clearly can introduce Type I errors. Emphasizing the effect on one gene or range of CpG sites obscures the fact that a network of genes are most likely turned on or off by the exposure. Candidate epigenetic approaches stemming from the animal literature are attractive because they demonstrate the importance of how a single gene may be causally implicated in a behavioral outcome. However, we should not be seduced by these elegant studies when trying to translate the animal research to humans because it is impossible to gain comparable experimental control.
Furthermore, we risk being shortsighted with respect to the epigenetic influences on behavior, due to an overemphasis in the published literature of the effects of a handful of genes on a child phenotype of interest (e.g., NR3c1, 11β-HSD2, and FKBP5). This is concerning given recent evidence that effect sizes are small and often fail correction for multiple comparisons (Breton et al., Reference Breton, Marsit, Faustman, Nadeau, Goodrich, Dolinoy and Murphy2017; Mansell et al., Reference Mansell, Vuillermin, Ponsonby, Collier, Saffery and Ryan2016). Another disadvantage is that we typically select a candidate gene because it is involved in a known physiological or biochemical process (Zhu & Zhao, Reference Zhu and Zhao2007). Yet we have a limited understanding of the biological etiology of complex phenotypes, such as mood disorders, and because of this we may be ignoring a network of genes that play a role in the prenatal origins of depression and other psychopathology. Finally, many candidate epigenetic studies fail to account for cellular heterogeneity. Within any given tissue, including brain and blood, there are many different types of cells, and high levels of DNA methylation found in a particular tissue may be due to the large numbers of particular kinds of cells, such as T cells, found in the tissue of one individual over another. For instance, candidate DNA methylation studies published using blood samples may actually be confounded by blood heterogeneity between individuals (de Goede et al., Reference de Goede, Razzaghian, Price, Jones, Kobor, Robinson and Lavoie2015). Although there are statistical methods to control for cell type in blood, buccal, and placental samples (Houseman et al., Reference Houseman, Accomando, Koestler, Christensen, Marsit, Nelson and Kelsey2012), these controls are rarely used in developmental behavioral epigenetic research (for an exception, see Paquette et al., Reference Paquette, Houseman, Green, Lesseur, Armstrong, Lester and Marsit2016).
Of 13 studies identified through a PubMed search on associations between an epigenetic mechanism and fetal or newborn neurobehavior, 11 utilized a candidate gene approach (Table 1; 7/13 focused on NR3c1 and 6/13 on HSD11β2). Seven examined how DNA methylation of NR3c1 was related to individual differences in newborn neurobehavior. Greater DNA methylation of NR3c1 likely impacts altered glucocorticoid receptor gene expression in the hippocampus, resulting in greater circulating cortisol in the blood (Meaney, Reference Meaney2010). DNA methylation of NR3c1 has been associated with infant movement and attention (Bromer, Marsit, Armstrong, Padbury, & Lester, Reference Bromer, Marsit, Armstrong, Padbury and Lester2013), and infant attention, self-regulation, lethargy, and soothability in a group of newborns exposed to maternal smoking in utero (Stroud et al., Reference Stroud, Papandonatos, Salisbury, Phipps, Huestis, Niaura and Lester2016). Interactions between NR3c1 and maternal depression are related to newborn self-regulation, lethargy, and hypotonia (Conradt, Lester, Appleton, Armstrong, & Marsit, Reference Conradt, Lester, Appleton, Armstrong and Marsit2013).
Table 1. Studies examining associations between epigenetic processes and fetal or newborn neurobehavior

HSD11β2 may regulate the extent to which the fetus is exposed to maternal cortisol given that this gene functions to convert maternal cortisol to inert cortisone. Greater methylation of this gene should therefore result in increased fetal cortisol exposure. Monk et al. (Reference Monk, Feng, Lee, Krupska, Champagne and Tycko2016) found that higher levels of maternal stress in the second trimester was related to greater methylation of placental HSD11β2 methylation and, in turn, decreased fetal coupling in the third trimester, thought to reflect impaired central nervous system development. DNA methylation of HSD11β2 has also been associated with quality of movement (Marsit, Maccani, Padbury, & Lester, 2012) and to a reactive, poorly regulated newborn neurobehavioral profile (Paquette et al., Reference Paquette, Lester, Lesseur, Armstrong, Guerin, Appleton and Marsit2015). Interactions between HSD11β2 and NR3c1 methylation were associated with habituation, excitability, and poor reflexes (Appleton, Lester, Armstrong, Lesseur, & Marsit, Reference Appleton, Lester, Armstrong, Lesseur and Marsit2015). These findings converge on the premise that exposure to maternal mood disorder may result in increased methylation of placental genes that expose the fetus to greater levels of cortisol in utero, which may then impact the infant's neuroendocrine system and newborn–child neurobehavior.
EWAS: Advantages and Disadvantages
EWAS is the emerging dominant analytical paradigm in behavioral epigenetics, and is apt to continue its rapid expansion given significant advantages over candidate epigenetic approaches. Its principle advantage lies in the ability to investigate the entirety of the epigenome in an unbiased fashion, the vast majority of which is poorly characterized and of unknown function (Swedish Schizophrenia Consortium et al., Reference McClay, Shabalin, Dozmorov, Adkins, Kumar and van den Oord2015). Such comprehensive epigenomic approaches are critical given our limited understanding of both the neurobiological mechanisms underlying human psychopathology and the epigenetic mechanisms regulating gene expression (Szyf, Reference Szyf2012). As the effects of prenatal exposures likely affect multiple biological systems and various functionally relevant genomic elements, an optimal approach must be comprehensive (Maccari, Krugers, Morley-Fletcher, Szyf, & Brunton, Reference Maccari, Krugers, Morley-Fletcher, Szyf and Brunton2014). Many current EWAS methylation assays satisfy these criteria (Adkins et al., Reference Adkins, Rasmussen, Docherty and Hopcroft2017).
A growing body of research has applied EWAS methylation approaches to study psychopathology, typically using array-based methods, which are standardized chips assaying a limited number of specific methylation loci (typically ≲ 850K CpG sites; Bock, Reference Bock2012). Fundamentally, such EWAS approaches are analytically agnostic, as they aim to unbiasedly assay the entire methylome. Thus, EWAS is liberated from the narrow restriction of looking only under the lampposts of existing biological knowledge, a fundamental limitation of candidate gene methods (Adkins et al., Reference Adkins, Rasmussen, Docherty and Hopcroft2017). EWAS is also critical to hypothesis generation as an inductive discovery tool, the findings of which can then be further refined in deductive, targeted epigenetic analyses. In addition, while most epigenetic studies of mood disorders have used candidate gene approaches, several EWAS, using a variety of technologies and examining hundreds of thousands of methylation markers across a range of tissues, have also been reported (Labonté et al., Reference Labonté, Suderman, Maussion, Navaro, Yerko, Mahar and Turecki2012, Reference Labonté, Suderman, Maussion, Lopez, Navarro-Sánchez, Yerko and Turecki2013; Nagy et al., Reference Nagy, Suderman, Yang, Szyf, Mechawar, Ernst and Turecki2015; Weder et al., Reference Weder, Zhang, Jensen, Yang, Simen, Jackowski and Kaufman2014). For example, EWAS have implicated several putative risk methylation marks for mood disorders, including loci at CPSF3, LASS2, PRIMA1, ZNF263, ID3, TPPP, GRIN1, STK32C, and ZBTB20 (Davies et al., Reference Davies, Krause, Bell, Gao, Ward, Wu and Wang2014; Dempster et al., Reference Dempster, Wong, Lester, Burrage, Gregory, Mill and Eley2014; Weder et al., Reference Weder, Zhang, Jensen, Yang, Simen, Jackowski and Kaufman2014).
EWAS is, however, not without its disadvantages. EWAS is frequently severely underpowered due to a combination of small sample sizes, small effect sizes, and punitive multiple test corrections (Tsai & Bell, Reference Tsai and Bell2015). In addition, while the cost of EWAS has dropped significantly, assays are still relatively expensive and large samples remain rare (Breton et al., Reference Breton, Marsit, Faustman, Nadeau, Goodrich, Dolinoy and Murphy2017). Moreover, the analytical problem of multiple testing in massively dimensional contexts has only increased as assay sizes have increased from 450K arrays, to 850K arrays, to next-generation sequencing methods assaying tens of millions of sites (Adkins et al., Reference Adkins, Rasmussen, Docherty and Hopcroft2017). Relatedly, EWAS is further limited by its failure to take advantage of the massive amounts of high-quality, prior information on genomic elements and biological pathways that is freely available through open-access bioinformatics databases (Huber et al., Reference Huber, Carey, Gentleman, Anders, Carlson, Carvalho and Morgan2015).
While accounting for correlated tests using false discovery rates can substantially increase power compared to Bonferroni correction (Benjamini & Hochberg, Reference Benjamini and Hochberg1995; Swedish Schizophrenia Consortium et al., Reference McClay, Shabalin, Dozmorov, Adkins, Kumar and van den Oord2015), it remains the case that effect sizes must be large in order to be detected in methylome-wide contexts (i.e., p ≲ 5 × 10−6). Unfortunately, effect sizes in EWAS, similar to genome-wide association studies, tend to be small (Breton et al., Reference Breton, Marsit, Faustman, Nadeau, Goodrich, Dolinoy and Murphy2017), necessitating large samples to robustly detect individual effects. For these reasons, to date, EWAS has suffered replication problems, particularly in psychiatry, where few findings have been replicated in large independent samples (McCarthy et al., Reference McCarthy, Abecasis, Cardon, Goldstein, Little, Ioannidis and Hirschhorn2008). In addition to limitations in statistical power, the agnosticism of the EWAS approach also limits the approach by ignoring the immense body of high-quality prior information that is publicly available in bioinformatics databases and published summary statistics. This has prevented EWAS approaches from boosting power through aggregating individual small effects into functional pathways and polygenic risk scores (de Moor et al., Reference de Moor, van den Berg, Verweij, Krueger, Luciano, Arias Vasquez and Boomsma2015; Docherty, Moscati, Dick, et al., Reference Docherty, Moscati, Dick, Savage, Salvatore, Cooke and Kendler2017), while also limiting our ability to extract mechanistic biological inferences from EWAS findings.
Two out of the 13 studies testing epigenetic–neurobehavioral associations used EWAS (Maccani et al., Reference Maccani, Koestler, Lester, Houseman, Armstrong, Kelsey and Marsit2015; Paquette et al., Reference Paquette, Houseman, Green, Lesseur, Armstrong, Lester and Marsit2016). Maccani et al. found that DNA methylation differences between newborns who were characterized as “high risk” due to their poor regulation; 6 out of 19 of these loci came from a gene EMID2, which is implicated in asthma and neural tube patterning. Paquette et al. found that DNA methylation of FHIT and ANKRD11, associated with brain and neurobehavioral development and placental physiology, were related to infant attention. These EWAS studies are likely to yield novel genes and genetic regions associated with newborn neurobehavior. However, it is not clear why some of these genes may be involved in neurobehavioral development. We turn now to a description of the functional epigenetic pathway that leverages the strengths of the candidate epigenetic and EWAS methods to interrogate epigenetic alterations in a known biological pathway of interest.
Functional Epigenetic Pathway Analyses
We argue here for a hybrid candidate epigenetic/EWAS approach that we term functional epigenetic pathway. This approach capitalizes on the advantages of the candidate epigenetic and EWAS techniques while minimizing their disadvantages. It is theory-driven and utilizes state-of-the art bioinformatic techniques to provide mechanistic insights into how an exposure may be related to an epigenetic phenomenon and phenotypic outcome. We should emphasize that while most of the literature to date has examined associations between maternal mood disorders, genes that regulate HPA axis functioning and fetal and newborn neurobehavior, the physiological data linking maternal mood and HPA axis functioning have been mixed.
A functional epigenetic pathway may advance the science of prenatal programming research in two ways: first, by targeting a known biological pathway of interest, and second by examining transcription factor binding sites. It is important to target transcription factor binding sites because of their role in gene expression. DNA methylation can lead to the silencing of the anticipated activity of a gene but only if DNA methylation occurs close to transcription factor binding sites. Therefore, this approach responds to recent concerns raised about the definition of the term epigenetic since this functional approach assumes that the phenotype of interest is mediated by DNA methylation at transcription factor binding sites (Greally, Reference Greally2018). Given their importance for regulating gene expression, it is surprising that these regions are typically not targeted in behavioral epigenetic research. Of the 13 studies reviewed investigating the possible epigenetic basis for effects of prenatal exposure to maternal mood disorder on fetal and newborn neurobehavior, 2 focus on these transcription factor binding sites (see Table 1). This may be because the field of behavioral epigenetics is in its infancy, and initial investigations focused on finding any possible associations between a prenatal exposure, DNA methylation, and a behavioral outcome. We argue that to advance the field of behavioral epigenetics, it is important to target DNA methylation of transcription factor binding sites, as opposed to other regions on the gene that may not have any functional significance, with the important caveat that, in the absence of tests of this mediation at the cellular level, we cannot infer direction of effect (e.g., that DNA methylation ![]() transcription factor binding or vice-versa; Lappalainen & Greally, Reference Lappalainen and Greally2017).
transcription factor binding or vice-versa; Lappalainen & Greally, Reference Lappalainen and Greally2017).
Our goal is to redirect the field of prenatal programing and behavioral epigenetics to focus on a functional epigenetic pathway approach targeting the HPA axis. This would be accomplished first through a comprehensive literature review identifying published associations between genes and the biological pathway of interest. This theory-driven approach adds specificity to current prenatal programming theories (Nigg, Reference Nigg2016) by targeting epigenetics marks along a hypothesized biological pathway of interest (in this case, the HPA axis). There are also public pathway databases available (e.g., the Broad Institute's Molecular Signatures Database v6.0 (MSigDB; Subramanian et al., Reference Subramanian, Tamayo, Mootha, Mukherjee, Ebert, Gillette and Mesirov2005) that allow researchers to identify coregulated gene networks in, for example, genes associated with the HPA axis. Following the identification of relevant genes, EWAS can be conducted that could generate additional novel sites associated with the biological pathway of interest. This information can be pooled together to generate a long list of specific genes and gene regions that, after controlling for multiple comparison, are associated with the biological pathway, termed the functional epigenetic pathway. The functional epigenetic pathway should then be replicated in an independent sample. If successful, this pathway could generate novel therapeutic targets for mitigating the risk of prenatal exposure to maternal mood on neurobehavioral development.
It is also possible to take a polygenic risk score approach to identifying which CpG sites may have strong associations with either prenatal exposure to maternal mood disorder or newborn neurobehavior. While the polygenic risk score approach has recently become enormously popular and productive in the analysis of genome-wide association studies genotype data (Gandal, Leppa, Won, Parikshak, & Geschwind, Reference Gandal, Leppa, Won, Parikshak and Geschwind2016), there has been very little application of the approach to genome-wide methylation data. Thus, polygenic risk score holds tremendous promise for epigenetic research, and is currently limited only by the size of existing training data (e.g., prior EWAS results for maternal mood). This approach leverages techniques used in bioinformatics that are already being applied in cancer research and molecular biology, but have yet to be applied to the study of human behavior.
Limitations and Conclusions
In this paper we have introduced a novel approach to studying epigenetic processes. The functional epigenetic pathway method uses bioinformatics to identify epigenetic markers empirically rather than using traditional candidate epigenetic or EWAS methods. This technique capitalizes on the strengths of both the candidate epigenetic and EWAS methods by being theory driven while also opening up the exploration process to discover larger epigenetic networks. Such networks will be functionally related and associated with known biological mechanisms underlying problematic newborn neurobehavior. However, it is also possible that novel pathways or interconnections will be uncovered with this technique, enriching our understanding of biological underpinnings of complex phenotypes. The functional epigenetic pathway approach proposed here builds upon similar work in the genetics literature, which has also increasingly applied bioinformatics to uncover gene networks.
Our novel approach to investigating how epigenetic processes may be implicated in prenatal exposure to maternal mood disorder is not without its limitations. First, it is extremely difficult to ascertain direction of effect in human behavioral epigenetic studies and any associations between DNA methylation and a phenotype of interest should be tested using cellular models that test for cellular reprogramming as a consequence of the exposure of interest (Lappalainen & Greally, Reference Lappalainen and Greally2017). Second, we begin by proposing only one biological pathway, the HPA axis, which has received a disproportionate amount of attention in the early life stress literature. There are mechanisms other than the HPA axis that should also be interrogated using a functional epigenetic pathway approach. A full review of additional stress response systems and pathways is beyond the scope of this paper, but the immune system and inflammation, serotonin system, imprinted genes, and mitochondrial biology will likely yield additional insights as to how prenatal maternal mood disorder exposure may alter the fetal epigenome and affect future child health.
Third, we have documented here that in the prenatal stress literature, there is an overreliance on the HPA axis as a mechanism by which prenatal exposure to maternal mood disorder may impact newborn neurobehavioral health that has yielded inconsistent results. Nevertheless, we are proposing to interrogate a network of genes implicated in HPA axis functioning to determine if methylation of these genes could explain the emergence of newborn neurobehavioral and downstream psychiatric problems. We argue that the reason for the mixed findings may be because we have not used the integrated functional epigenetic assay approach, which is hypothesis driven and leverages a key biological pathway that is implicated in some manner in research on maternal mood disorders. By testing the validity of this assay, we may ultimately be able to determine whether the HPA axis is a key mediator linking prenatal stress with impaired newborn neurobehavioral and gestational health (e.g., preterm birth) outcomes.
Fourth, we propose to examine epigenetic effects in tissues other than the brain, such as the placenta, buccal cells, or blood. There is no established protocol for placenta collection, which is limited by the fact that placental RNA quality is affected by processing time postdelivery. In addition, there are disagreements as to whether cord blood should be used for DNA methylation analyses because the presence of nucleated red blood cells could contaminate cell populations and skew DNA methylation profiles. However, the presence of these disparate cell types can be accounted for in post hoc statistical analyses. Single-cell sequencing can also be used to characterize different cell types (e.g., scRNA-seq; Lappalainen & Greally, Reference Lappalainen and Greally2017). Relatedly, placenta and blood samples include a mixture of different cell types, though this concern can also be raised when studying brain tissue. Nevertheless, if we hope to incorporate epigenetic processes as biomarkers for pathophysiological processes related to HPA axis development in the fetus, peripheral tissues must be used.
Fifth, we described this approach in the context of maternal mood disorder, which has also received a disproportionate amount of attention in the epigenetic literature (Conradt, Reference Conradt2017). It is important to note that depression is a highly heterogeneous diagnosis. In addition to symptom-level heterogeneity (i.e., the fact that people can meet diagnostic threshold via unique combinations of symptoms), there are many distinct etiological pathways leading to depression and different comorbidities. For example, it is possible that depression in the context of antisocial personality disorder would have a different effect on child outcomes than depression in the context of anxiety. Depressive symptoms can also arise secondary to life stress, health problems, and any number of other life circumstances. Future epigenetic research should focus less on specific psychiatric diagnoses and instead emphasize broad trait-level vulnerabilities, such as emotion dysregulation or impulsivity. Such traits may be more closely linked to genetic and epigenetic processes relative to complex distal phenotypes such as depression.
We believe that testing this novel functional epigenetic pathway has potential to benefit public health and the field of behavioral epigenetics. Particularly exciting to us is the possibility that other researchers will apply this pathway approach to their own research questions that implicate the HPA axis in the expression of disorder. For example, there are a number of additional stress and immune-related pathways that could be also be interrogated using the methods described above. Furthermore, the use of tools, such as epigenetic aging studies, could bring specificity to how prenatal experiences become biologically embedded. Ultimately, we expect that the application of this functional epigenetic network approach will improve prevention efforts aimed at reducing the effects of maternal mood exposure prenatally and could result in novel therapeutic targets to buffer the fetus against maternal mood dysregulation, stress, and other risk processes.


