



Introduction
Attention-deficit/hyperactivity disorder (ADHD) is a common neurodevelopmental disorder with an estimated worldwide prevalence of 4.4% to 5.2% in adults aged 18 to 44 yearsReference Kessler, Adler and Barkley 1 , Reference Fayyad, Sampson and Hwang 2 and 2.85% to 3.5% in adults aged 50 years or older.Reference Surman and Goodman 3 Prospective longitudinal studies have provided additional evidence that approximately 71% of individuals diagnosed as children continue to demonstrate symptoms of impairment well into adulthood.Reference Faraone, Asherson and Banaschewski 4 Interest in extended-release, longer-acting psychostimulants for the treatment of ADHD in adults has been driven by an increasing body of evidence in support of the use of amphetamine and methylphenidate for this patient population.
Amphetamine extended-release oral suspension (AMPH EROS; Dyanavel® XR, Tris Pharma, Inc., Monmouth Junction, NJ) was approved by the U.S. Food and Drug Administration (FDA) in 2015 for the treatment of ADHD. At initial approval, the product was approved for use in children 6 years of age and older. The labeled indication was later expanded to include adults. 5 The basis for this FDA-approved label change was a clinical bioavailability study comparing equal doses of AMPH EROS to an extended-release mixed amphetamine salt (MAS) product that is approved for use in adults with ADHD.
AMPH EROS is a scalemic amphetamine formulation that utilizes the proprietary LiquiXR® drug delivery technology. The LiquiXR® proprietary drug delivery technology utilizes an ion-exchange resin that complexes with amphetamine.Reference Herman, King, Kando and Pardo 6 The active drug product forms a complex with ion-exchange polymers contained in the resin, which is then formed into micron-sized particles. Some of these particles are coated with an aqueous, pH-independent polymer of varying thickness, allowing for programmed, extended release of active drug product. Solid, coating-free particles provide for immediate release of active drug product. Drug–resin complex particles either remain uncoated (facilitating immediate release of the drug product; in this case, amphetamine) or coated in a range of variable thicknesses (facilitating extended release of amphetamine). The micron-sized drug–resin complex particles are formulated into an appropriate dosage form (solid or chewable tablet, liquid suspension, orally disintegrating tablet, film, or capsules). After ingestion, amphetamine is subsequently released from the dosage form in millions of particles, with the release driven by a combination of ion exchange and diffusion. After amphetamine release, the ion-exchange resin is excreted in the feces.
A previous study described the PK (PK) of AMPH EROS in a healthy adult population. In that study, AMPH EROS was found to have comparable bioavailability in adults to an immediate-release formulation of mixed amphetamine salts.Reference Herman, Bouhajib, King, Kando and Pardo 7 To better define the PK of the test product AMPH EROS in adults, the purpose of the present study was to evaluate the relative bioavailability of a single dose of AMPH EROS compared with the reference product, a single dose of an extended-release mixed amphetamine salts (ER MAS) formulation, in healthy adult subjects under fasting conditions.
Methods
Study design
This was an open-label, single-dose, randomized, two-period, two-treatment, two-sequence, crossover, and comparative bioavailability study that was conducted at a single site (Pharma Medica Research, Inc., St. Charles, MO). The study was conducted in accordance with the principles of the U.S. Investigational New Drug regulations codified in the United States Code of Federal Regulations (21 CFR 312), and in compliance with International Council for Harmonization Guidelines for Good Clinical Practice 8 and the current U.S. FDA guidance document, Guidance for Industry-Bioavailability and Bioequivalence Studies for Orally Administered Drug Products. 9 All subjects provided written informed consent prior to engagement of any study procedures. The protocol and informed consent documentation were approved by an Institutional Review Board (Salus IRB, Austin TX).
Study subjects
The study population consisted of healthy, nonsmoking, male and female volunteers ages 18 to 55 years with a body mass index (BMI) ≥19.0 and ≤33.0 kg/m2. A general screening procedure was conducted and only volunteers that were considered healthy were eligible to participate in the study. Clinical laboratory tests administered at screening included a baseline complete blood count and blood chemistries. A complete medical history, blood pressure, pulse, and respiratory rate measurements were also obtained. Enrolled subjects met the inclusion criteria, did not fulfill any of the exclusion criteria, and satisfied subject selection criteria no more than 28 days prior to the first drug administration. Females of childbearing or nonchildbearing potential, including those who were sterile (ie, both ovaries removed, uterus removed or bilateral tubal ligation for at least 6 weeks or documented successful hysteroscopic sterilization); and/or postmenopausal (no menstrual period for at least 12 consecutive months without any other medical cause) were considered eligible to participate. Females were instructed to use an acceptable single- or double-method of contraception from 21 days prior to drug administration until 28 days after the last PK blood sample in the study. Males were instructed to use an acceptable single- or double-method of contraception from the day of drug administration until 28 days after the last PK blood sample in the study. All eligible subjects were determined to be able to tolerate venipuncture and were able to fully comprehend the nature of the study and give written consent prior to any study procedure.
Key exclusion criteria included (but were not limited to) a known history or presence of clinically significant cardiovascular, neurologic, or psychiatric condition which, in the opinion of the investigator, would jeopardize the safety of the subject or impact the validity of the study results.
The study drug/test product used in this study was 7.5 mL of 2.5 mg/mL AMPH EROS (DYANAVEL® XR, Tris Pharma, Inc., Monmouth Junction, NJ) equivalent to 18.8 mg of amphetamine base administered after an overnight fast of at least 10 hours. The AMPH EROS used in this study comprises a 3.2:1 enantiomeric ratio of d-amphetamine to l-amphetamine. The reference product was one 30 mg ER MAS capsule (ADDERALL XR®, Shire USA Inc., Wayne, PA) equivalent to 18.8 mg of amphetamine base, also administered after an overnight fast of at least 10 hours.
PK sampling
A catheter for venous sampling was placed. Blood samples were collected prior to dosing (0-hour) and at 1, 2, 3, 3.5, 4, 4.5, 5, 6, 7, 8, 9, 10, 12, 14, 16, 24, 36, 48, and 60 hours after drug administration, totaling 20 samples in each period. Postdose blood samples were collected in prechilled, labeled, 4 mL blood collection tubes containing K2EDTA as the anticoagulant. Approximately 170 mL of blood was collected, by direct venipuncture, from each subject, over the entire study for PK analysis. The plasma was subsequently divided into two equal aliquots in polypropylene tubes and stored frozen at −25°C ±10 until shipment for analysis at the analytical laboratory (Pharma Medica Research Inc., Bioanalytical Division, Ontario, Canada). The sampling schedule was designed for this study to ensure that the area under the curve (AUC) AUC and maximum concentration (Cmax) C max parameters were adequately and safely assessed from collected plasma. By implementing a crossover design, the estimated PK parameters for each product were compared within the same subject. The crossover design allowed for intra-subject PK comparisons. To prevent any carryover effect, the two study periods were separated by a washout of at least 7 days, corresponding to a time interval equivalent to more than five times the expected drug plasma elimination half-lives of d-amphetamine and l-amphetamine.
Relative bioavailability between the marketed products was determined by a statistical comparison of the AUC and C max parameters for d-amphetamine and l-amphetamine. Partial AUCs were used to better characterize the biphasic release components of the extended-release formulations tested.
Subjects were randomly assigned to one treatment sequence, according to a predetermined computer-generated randomization scheme (procedure PLAN in SAS®): Sequence 1: test product AMPH EROS then reference ER MAS, and Sequence 2: ER MAS followed by AMPH EROS. Subjects were assigned consecutive numbers in an ascending order. Each number identified a subject and determined the sequence of drug product administration according to the randomization scheme.
All prescription or over-the-counter medications were to be discontinued 14 days prior to administration of study drug. Exceptions were made for: hormonal contraceptives; nonsystemic and/or topically applied products (prescription or otherwise); and occasional use of common analgesics. Subjects were sequestered in-house from at least 10 hours prior to drug administration until at least 60 hours postdose in each period. Subjects fasted from for at least 10 hours prior to drug administration until at least 4 hours postdose. Standardized meals were provided throughout the in-house study component. The meals were identical for both periods. Water was restricted from one hour prior to drug administration until 1-hour postdose (except the water administered with the drug). Access to water was otherwise freely available to subjects.
Qualified clinic staff ensured that all study drugs were administered according to the protocol and randomization scheme. Prior to drug administration, subjects were instructed not to touch, swirl, or spit out any of the study drug. Subjects who touched, swirled, or spat out the study drug were to be removed from the study.
A single dose of active drug product 7.5 mL of 2.5 mg/mL (equivalent to 18.8 mg amphetamine base) of AMPH EROS was administered according to the randomization scheme followed by 240 ± 5 mL of room temperature potable water. For AMPH EROS administration, each subject was in a seated position with their head tilted slightly upwards. The oral syringe was placed into the subject’s mouth at an angle and was facing downwards. The upper portion of the oral syringe (not just the tip) was inserted into the subject’s mouth and the subjects were asked to close their mouths tightly around the oral syringe. At the time of drug administration, the drug was released directly into the subject’s mouth in one continuous push. The subjects were asked to swallow all of the suspension in their mouth but were permitted to swallow in portions if desired (with the syringe remaining in the subject’s mouth). Each subject then consumed the room temperature potable water, by lightly “swishing” the water in their mouth, and then swallowing. All attempts were made to administer AMPH EROS and dosing water within 2 minutes. The beginning of the release of the syringe’s contents into the subject’s mouth was recorded as the dosing time.
For administration of the reference product, the single 30 mg ER MAS capsule was administered, followed by consumption of the room temperature potable water, by lightly “swishing” the water in their mouth, and then swallowing. A hand and mouth check was performed immediately after drug administration, in both treatments, to ensure that the study drug had been swallowed.
Subjects remained seated for 4 hours following drug administration (unless required to ambulate for study-specific procedures or use the restroom) and were permitted to resume normal activity thereafter. Subjects did not engage in any strenuous activity and were permitted to lie down if they experienced drowsiness, dizziness, or other AEs requiring such a position.
Analytical methods
Plasma concentrations of l- and d-amphetamine in subject samples were measured using a chiral, liquid chromatographic tandem mass spectrometric detection method (LC-MS/MS) developed and validated at the Bioanalytical Laboratory of Pharma Medica Research Inc (Mississauga, ON, Canada). The standard calibration range for the method was from 0.200 to 80.0 ng/mL for each enantiomer using a plasma sample volume of 0.100 mL. Plasma samples were extracted under alkaline conditions with an organic solvent; the organic phase was dried and reconstituted with buffer. Reconstituted samples were derivatized and further processed with a second liquid-liquid extraction prior to analysis by LC-MS/MS. Chromatographic separation of the enantiomers was achieved using a chiral column (100 × 4 mm, 5 μm). Derivatized l- and d-amphetamine were analyzed in the SCIEX API 4000 mass spectrometer using positive ion scan mode with a parent–daughter mass to charge ion transition of 418-91. Similarly, the derivatized internal standards, l-amphetamine-d10 and d-amphetamine-d10, were analyzed using a parent–daughter mass to charge transition of 428-97. The expected retention time for l-amphetamine and its internal standard was approximately 3.5 minutes and the retention time of d-amphetamine and its internal standard was approximately 3.9 minutes. Calibration standards were prepared by spiking blank human plasma with known amounts of racemic amphetamine to create one set of nine nonzero calibration standards (0.200, 0.400, 1.00, 2.50, 5.00, 12.5, 25.0, 50.0, and 80.0 ng/mL for each enantiomer). To assess method performance, quality control (QC) samples containing known amounts of racemic amphetamine were prepared: 0.600, 40.0, 65.0, and 7.50 ng/mL for each enantiomer. The back-calculated concentration of the lower limit of quantitation (0.200 ng/mL) was within ±20.0% of the nominal value and the back-calculated concentrations of all other calibration standards were within ±15.0% of their nominal values. A minimum of two-thirds (2/3) of the quality control samples and 50% at each concentration level were to be within ±15.0% of their respective nominal values. Plasma concentrations of l- and d-amphetamine in subject samples were measured using Analyst® Software Version 1.6.2 (AB Sciex PTE, Ltd., Singapore).
Statistics
Descriptive statistics for the PK parameters of d-amphetamine and l-amphetamine were calculated. Descriptive statistics included number of observations, arithmetic mean, standard deviation, geometric mean (where applicable), coefficient of variation (CV), median, minimum, and maximum. Analysis of variance (ANOVA) was performed on log-transformed AUC0-4, AUC4-t, AUC0-5, AUC5-t, AUCt, AUCinf, and C max parameters. The significance of the sequence, period, treatment, and subject (sequence) effects was tested.
Using the same statistical model, the least-squares-means (LSMs), the between-treatment differences between the treatment LSMs and the corresponding standard errors of these differences were estimated for log-transformed AUC0-4, AUC4-t, AUC0-5, AUC5-t, AUCt, AUCinf, and C max parameters. Based on these statistics, the ratios of the geometric means for treatments and the corresponding 90% confidence intervals (CIs) were calculated. In order to declare bioequivalence of AMPH EROS to the ER MAS capsule, the 90% CIs of the ratios of geometric mean plasma d-amphetamine and l-amphetamine AUC0-5, AUC5-t, AUCinf, and C max of the AMPH EROS in relation to the ER MAS capsule should be between 80.00% and 125.00%, inclusive.
In-house data indicated a CV for l-amphetamine AUC of approximately 21%. Assuming a 21% intra-subject variability and a difference between the treatment means of 5% or less, the necessary sample size for a 90% probability of the 90% CI of the treatment means ratio to be within the 80.00% to 125.00% range was estimated to be 26 subjects. Four extra subjects were included into the study to account for potential dropouts. Therefore, 30 subjects were enrolled into this study. Only volunteers who were dosed with test article or reference product were considered enrolled.
Safety
Subjects vital signs were continually monitored throughout the conduct of the study. Any adverse events occurring during study conduct were recorded.
Results
Study subjects
Thirty subjects were enrolled in the study and 28 completed the study. Twenty-nine subjects received AMPH EROS and all subjects received the ER MAS. One subject discontinued due to personal reasons, and a second subject was removed from the study due to a concomitant medication violation (odansetron). Demographics and baseline characteristics are provided in Table 1.
Table 1. Subject Demographics and Baseline Characteristics

Abbreviations: BMI, body mass index; N, number of subjects included in each dataset; n, number of subjects in respective parameters; SD, standard deviation.
PK assessments
In order to declare bioequivalence of the AMPH EROS to the ER MAS capsule, the 90% CIs of the ratios of geometric mean plasma d-amphetamine and l-amphetamine AUC0-5, AUC5‑t, AUCinf, and C max of AMPH EROS in relation to the ER MAS capsule were required to be between 80.00% and 125.00%, inclusive. This study yielded sufficient data to afford a view of the overall PK profile of AMPH EROS in this subject population of healthy adults. Quantifiable plasma concentrations of l- and d-amphetamine from AMPH EROS and ER MAS were detected as early as 1-hour postdose and subsequently throughout the entire 60-hour testing timeframe. A single high maximum concentration peak for AMPH EROS was noted around 3 to 4 hours postdose, with a single peak for ER MAS occurring slightly later, at approximately 6 hours postdose. A gradual descent in the curve was noted from the peak to the end of the sampling time frame for both the test article AMPH EROS and reference ER MAS. Similar profiles and PK characteristics were demonstrated between the l-amphetamine curves for AMPH EROS and ER MAS (Figures 1 and Table 2). The median T max for measurements for AMPH EROS were 4.00 and 4.02 hours for d- and l-amphetamine, respectively; and for ER MAS were 5.00 hours for d- and l-amphetamine, respectively.
Table 2. Geometric Mean Exposure of d- and l-Amphetamine
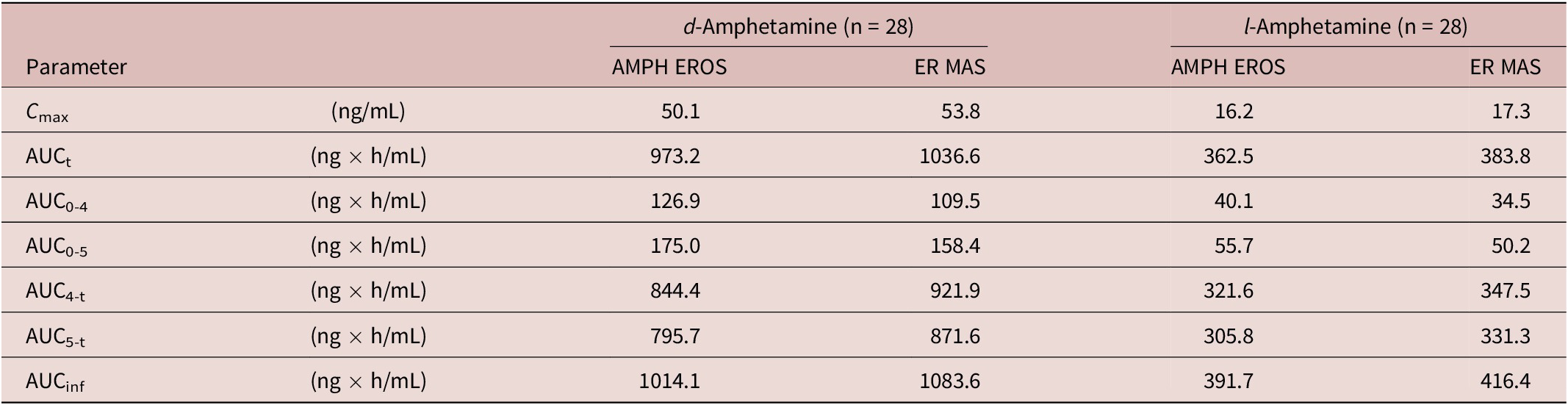
Abbreviations: AMPH EROS, amphetamine extended-release oral suspension; ER MAS, extended-release mixed amphetamine salts n = number of subjects in PK Dataset.
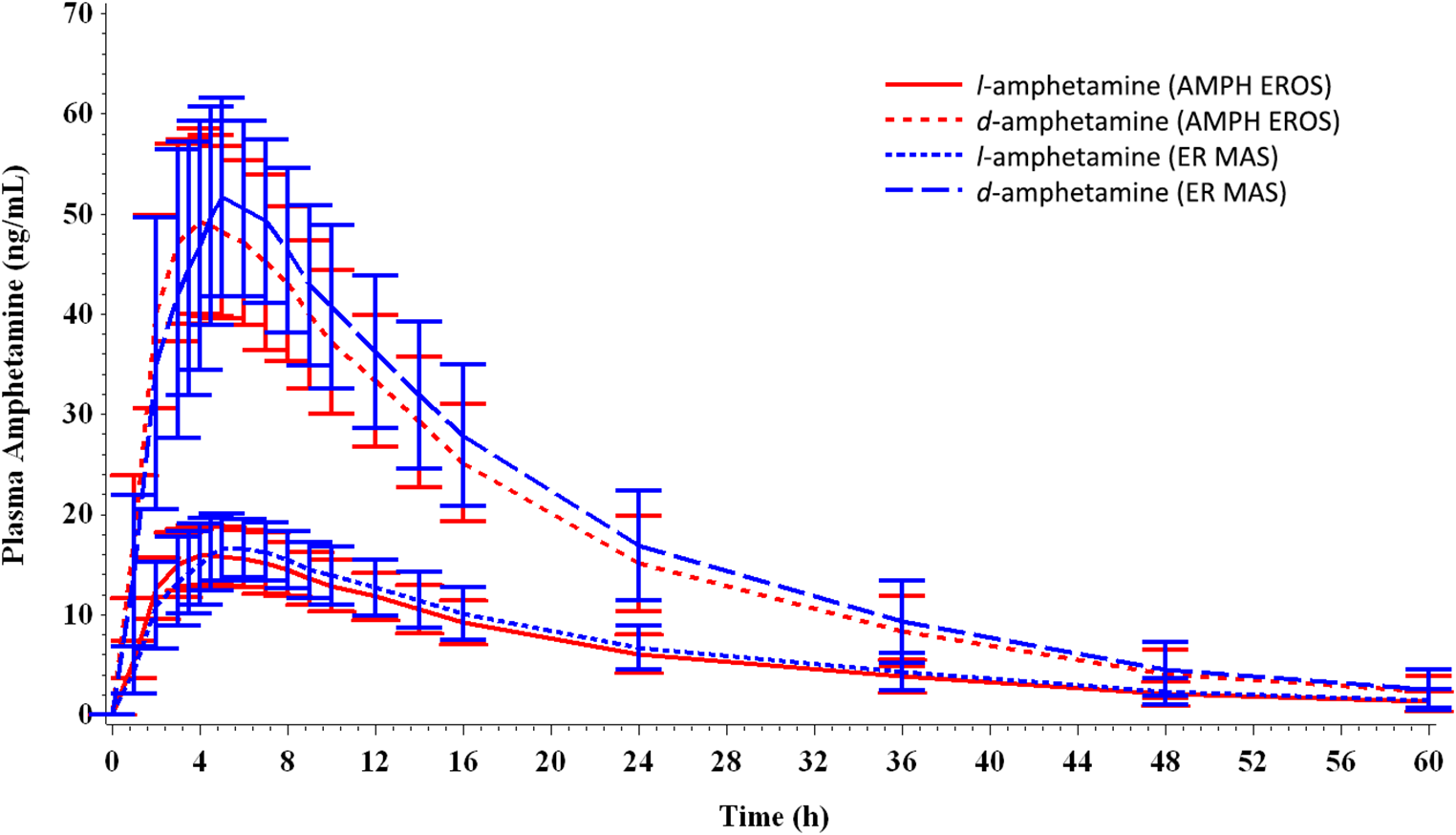
Figure 1. Mean plasma concentrations over time of d- and l-amphetamine (amphetamine extended-release oral suspension [AMPH EROS] and extended-release mixed amphetamine salts [ER MAS]).
Comparative bioavailability
As detailed in Table 3, the contrasts for geometric mean ratios for all assessed PK parameters (for both l- and d-amphetamine) between the test article AMPH EROS and reference product ER MAS fell within the prescribed 80% to 125% limits. The ANOVA did not detect a significant difference in any of the PK parameters for period and sequence effects.
Table 3. 90% Confidence Intervals of the Ratios of Geometric Means
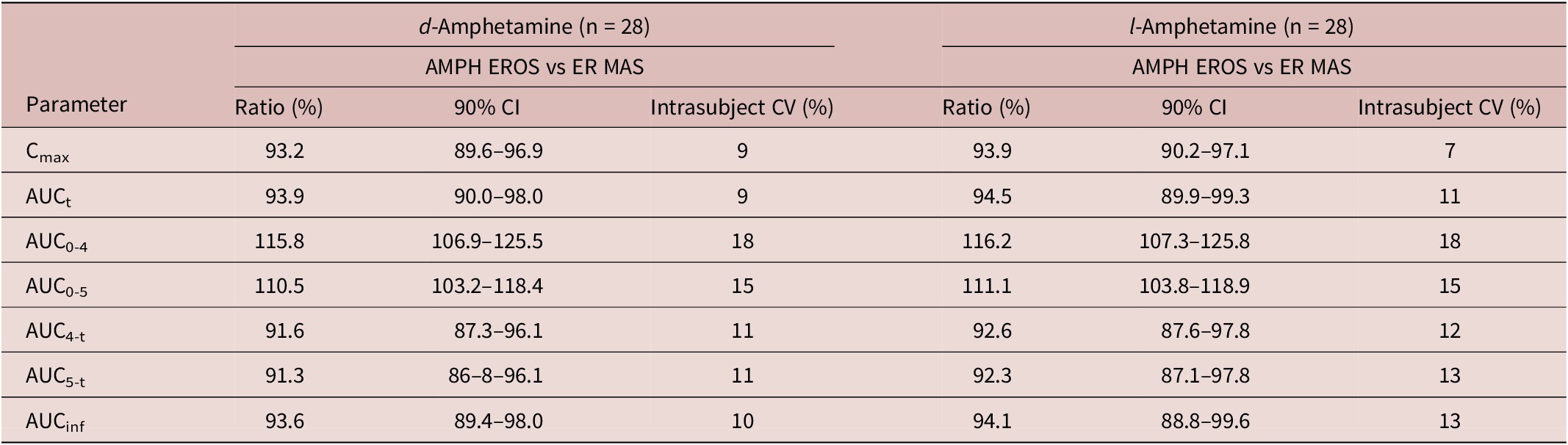
Abbreviations: AMPH EROS, amphetamine extended-release oral suspension; CI, confidence intervals; ER MAS, extended-release mixed amphetamine salts.
Safety
Twenty-nine subjects were dosed in both periods of the study and received an equivalent of 18.8 mg amphetamine base in each period, totaling 37.6 mg amphetamine base over the course of the study. A single subject discontinued from the study prior to period 2 dosing and was only administered a single dose of 18.8 mg amphetamine base.
The administration of the study drugs was generally well tolerated by the healthy subjects in this study. Overall, there were 23 TEAEs affecting 11 subjects (36.7% of all subjects who were dosed). Of these, 22 TEAEs in 10 subjects (33.3%) were deemed by the investigator to be treatment-related (possibly, probably, or definitely related). There were no unexpected significant findings related to vital signs, electrocardiograms or physical examinations in this study. No subjects discontinued from the study due to AEs and none of the AEs had a significant impact on the safety of the subjects.
Five subjects (17.2%) receiving AMPH EROS reported 5 treatment-related TEAEs and 7 subjects (23.3%) receiving ER MAS reported 17 treatment-related TEAEs. The most commonly reported treatment-related TEAE was tachycardia (6 events in 5 subjects [16.7%]). One subject experienced tachycardia after administration of AMPH EROS and four subjects experienced tachycardia after administration of ER MAS. No subjects reported tachycardia in both periods (or with either test article or reference product). Overall, the safety profile in this small population was consistent with the profile recognized for the drug class. 10 , 11
Discussion
In this study, the PK of the currently marketed formulation of an amphetamine extended-release oral suspension was compared with that of a marketed reference product extended-release mixed amphetamine salts capsule. AMPH EROS 7.5 mL of a 2.5 mg/mL solution (for 18.8 mg amphetamine base per 7.5 mL) exhibited equivalent total and peak exposure to ER MAS capsules, 30 mg in healthy adult subjects after a single, oral dose, under fasted conditions. Peak concentration (C max) was 93% and 94%, respectively, of the C max of ER MAS capsules. The relative bioavailability of AMPH EROS compared with an equal dose of ER MAS capsules is 94% for both d- and l-amphetamine. Peak plasma concentrations of both d-amphetamine and l-amphetamine occur approximately 3 to 5 hours following oral administration under fasting conditions. The comparative observations in this study involved a single dose and volume of each formulation.
In a previous PK study, following a single 18.8 mg oral dose of AMPH EROS in 29 healthy adult subjects under fasting conditions, the mean (±SD) plasma terminal elimination half-life of d-amphetamine was 12.36 (±2.95) hours and the mean (±SD) plasma terminal half-life for l-amphetamine was 15.12 (± 4.40) hours.Reference Herman, Bouhajib, King, Kando and Pardo 7 These results were noted in healthy adult subjects under fasting conditions, although the bioavailability of AMPH EROS at a dose of 18.8 mg in the presence of food has been studied and not shown to have any clinically important effect.Reference Herman, Bouhajib, King, Kando and Pardo 7 The PK data for AMPH EROS demonstrates a once-daily dosing PK profile with rapid absorption and peak concentration times, and a prolonged release of stimulant equivalent to the reference formulation ER MAS.
Conclusion
The overall PK profile of single-dose AMPH EROS 7.5 mL was found to be comparable with a single dose of ER MAS 30 mg administered orally. AMPH EROS and ER MAS were both well-tolerated in this study.
Acknowledgements
The authors thankfully acknowledge the study subjects for their participation. The authors also acknowledge Payal Naik, MPH, for her assistance in preparing this manuscript.
Disclosures
The study upon which this work was based was funded by Tris Pharma, Inc., the developer and manufacturer of amphetamine-extended release oral suspension. Drs. Pardo, Rafla, Kando, Mr. King, and Ms. Naik are paid employees of Tris Pharma, Inc. Dr. Kando holds stock in Takeda and Johnson and Johnson. Mr. King holds stock in Merck and Co, Inc., and Pfizer, Inc. Mr. Bouhajib (study PKist) is a paid employee of Pharma Medica Research, Inc., Mississauga, Ontario and was compensated for his work on the study. None of the authors hold any patents or copyrights broadly relevant to this manuscript. They report no other relationships or activities that readers could perceive to have influenced, or that give the appearance of potentially influencing what was written in this manuscript.




