In the framework of the Spanish programme on the deep geological disposal of high-level radioactive wastes, the Morrón de Mateo bentonite deposit has been studied as a natural analogue of the bentonite barrier behaviour after burial, in relation to the thermal and geochemical effects induced by the radioactive decay of the fission products and the interaction between the corrosion products from the steel canister and the bentonite, respectively. This bentonite deposit, including its host rocks and the overlying biocalcarenite beds, was intruded by a rhyodacitic volcanic dome which induced a metasomatic process by means of hydrothermal Fe-Mg-rich fluids, mainly manifested by the transformation of calcite from the biocalcarenite beds into Fe-Mg-rich dolomite in the zone nearest to the dome (Delgado, Reference Delgado1993; Pérez del Villar et al., Reference Pérez del Villar, Delgado, Reyes, Pelayo, Fernández-Soler, Cózar, Tsige and Quejido2005). Based on those results, the present authors carried out a mineralogical, chemical, geochemical and isotopic study of the smectites from the bentonite deposit in order to verify whether these bentonites were also affected by the aforementioned metasomatic process.Smectites located away from the dome were found to be dioctahedral Al-rich smectites, similar to those from other deposits in the region (Reyes, Reference Reyes1977; Caballero et al., Reference Caballero, Reyes, Yusta, Huertas and Linares1985; Delgado, Reference Delgado1993), while smectites located in the vicinity of the dome are a mixture of Al-montmorillonites, Fe-rich montmorillonites and beidellites, and intermediate smectites, between beidellite and Fe-rich saponite (Pelayo, Reference Pelayo2014; Pelayo et al., Reference Pelayo, García-Romero, Labajo and Pérez del Villar2016). Furthermore, the textural relationships observed by Scanning Electron Microscopy (SEM), coupled with an Energy Dispersive X-ray spectroscopy (EDX) system, suggested that the smectites with intermediate composition come from the gradual transformation of Al-montmorillonite to smectites with increasing Mg and Fe contents (Pelayo, Reference Pelayo2014; Pelayo et al., Reference Pelayo, García-Romero, Labajo and Pérez del Villar2016). To confirm this suggestion, smectites located distant from and close to the sub-volcanic dome were studied by infrared (FTIR) and Mössbauer spectroscopy to analyse the Fe distribution in the fine fractions of bentonite. The main goal of the present study was to understand and confirm the transformation processes that occurred in the smectites from the Morrón de Mateo deposit as a result of their interaction with the hydrothermal Fe-Mg-rich fluids related to the intrusion of the rhyodacitic volcanic dome.
Infrared spectroscopy is a very efficient method for studying the distribution of cations in the structure of phyllosilicates, especially in the octahedral sheet, as the OH bands are sensitive to both structural and compositional variations (Farmer, Reference Farmer1974; Madejová et al., Reference Madejová, Komadel and Číčel1994; Russell & Fraser, Reference Russell, Fraser and Wilson1994; Bishop et al., Reference Bishop, Madejová, Komadel and Froeschl2002). The IR region corresponding to both OH-stretching and bending vibrational modes provides qualitative and quantitative information about cation distribution in the octahedral sheet (Vantelon et al., Reference Vantelon, Pelletier, Michot, Barres and Thomas2001).
Mössbauer spectroscopy allows determination of the oxidation state of Fe in phyllosilicates, oxides and other Fe-rich minerals or admixtures, and, in favourable cases, also the Fe coordination, making this technique suitable for the characterization of their Fe-compounds (Coey, Reference Coey1980; Goodman, Reference Goodman and Wilson1994; Murad, Reference Murad2010). Additionally, because the quadrupole splitting of Fe(III) is determined by external charges, its magnitude provides a direct indication of the site distortion (Murad, Reference Murad2010). On the other hand, this spectroscopic technique allows distinction between Fe bound in oxides and in clay mineral structures, and also identification of the Fe-oxyhydroxide species present in the samples (Murad, Reference Murad, Stucki, Goodman and Schwertmann1988, Reference Murad2010; Komadel et al., Reference Komadel, Madejová and Stucki1999).
Furthermore, in laboratory experiments performed to study the stability of the bentonite barrier, as a consequence of its interaction with corrosion products of the Fe-rich container, both FTIR and Mössbauer spectroscopic techniques are usually employed to quantify the amount of the destabilized smectite, resulting from the interaction with Fe, which allows the comparison with the original smectite (Guillaume et al., Reference Guillaume, Neaman, Cathelineau, Mosser-Ruck, Peiffert, Abdelmoula, Dubessy, Villiéras and Michau2004; Lantenois et al., Reference Lantenois, Lanson, Muller, Bauer, Jullien and Plançon2005; Wilson et al., Reference Wilson, Cressey, Cressey, Cuadros, Ragnarsdottir, Savage and Shibata2006).
GEOLOGICAL SETTING
The Morrón de Mateo deposit is the main bentonite mass in the vicinity of the volcanic dome known as Morrón de Mateo. It is located in the central sector of the Cabo de Gata region (Almería), specifically in the Escullos depression, which is placed between the Frailes volcanic structure, to the south, and the Rodalquilar Caldera Complex, to the north. The deposit forms part of a Lower Tortonian volcano-sedimentary series (11.6 Ma), which mainly consists, from the bottom to the top, of the following geological units: (1) hornblende-rich andesitic breccias; (2) bentonitized pyroclastic layered rocks (White Tuffs formation); (3) marine sedimentary rocks; and (4) partially bentonitized epiclastic rocks (Mass Flow formation) (Fig. 1), which have been described extensively by Fernández-Soler (Reference Fernández-Soler2002). The Morrón de Mateo dome intruded through this volcano-sedimentary series. The White Tuffs formation is the main host and parent rock of the bentonite deposit and is composed of a succession of white-coloured, soft layers of bentonite-rich tuffs and tabular layers of a brownish to greenish sandy material, poorer in smectite, that form base surge and co-surge hydromagmatic facies. This formation shows some sedimentary structures that point to lateral currents as the main transport and deposition mechanism. These features suggest that the white-layered tuffs may have formed as a result of phreatomagmatic activity triggered by magma–water interaction in a shallow marine environment, probably in a depressed coastal embayment (Fernández-Soler, Reference Fernández-Soler1992).

Fig. 1. (a) Geological map of the Morrón de Mateo area with the studied boreholes (S2, S3, S4, SE-1, SE-3, SE-6, SE-8 and SE-10) and profile (P5) (modified after Fernández-Soler, Reference Fernández-Soler2002). (b) Geological cross-sections A–A' and B–B' showing the relationship between the sub-volcanic intrusion and the volcano-sedimentary sequence. Key: (1) hornblende-rich andesite breccias; (2) White Tuffs formation; (3) Tortonian biocalcarenite beds; (4) Mass flow formation; (5) Morrón de Mateo dome; (6) Quaternary sediments; (7) Rodalquilar complex rhyodacitic rock.
MATERIALS AND METHODS
Materials
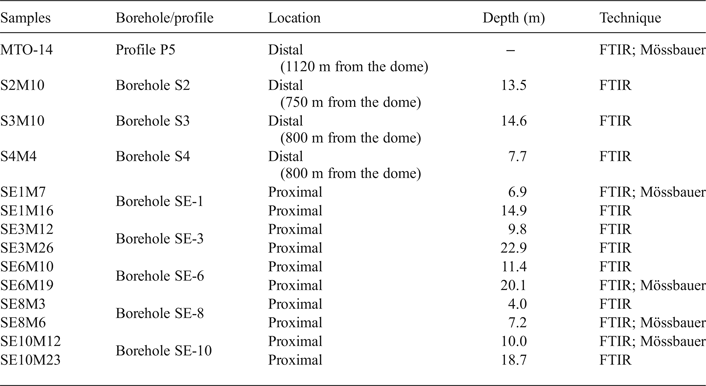
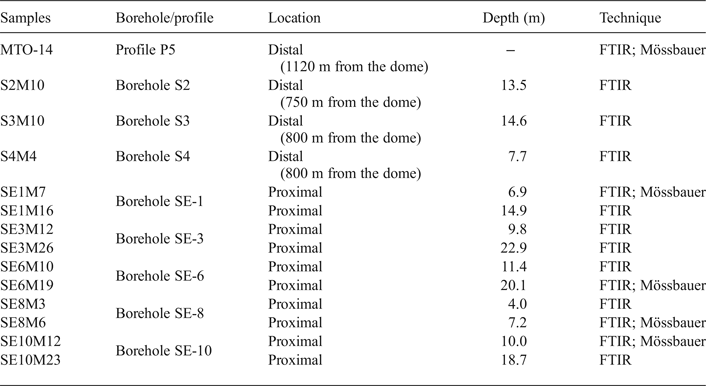
Fourteen representative samples from the White Tuffs Formation, previously characterized by X-ray diffraction (XRD) (Pelayo, Reference Pelayo2014), were selected for FTIR analysis. They were collected from eight boreholes. Three samples (S2, S3 and S4) are located away from the volcanic dome (distal samples), in the northeast zone of the Morrón de Mateo area, and the remaining samples (SE-1, SE-3, SE-6, SE-8 and SE-10) are located close to the dome (proximal samples), in the quarry to the south of the dome. The depth of the boreholes varies from 15 to 33 m. One other sample was taken from a surface cross-section (P5) located far from the dome, in the so-called El Murciano quarry (Fig. 1, Table 1). In addition, four samples from proximal boreholes (SE-1, SE-6, SE-8 and SE-10) and one sample from the distal surface profile (P5) were selected for Mössbauer spectroscopy (Table 1).
Table 1. Sample locations, depth and technique used in their study.
The < 2 µm fractions used in this study were from the White Tuffs formation, extracted by conventional sedimentation method (Moore & Reynolds, Reference Moore and Reynolds1997).
Sample description
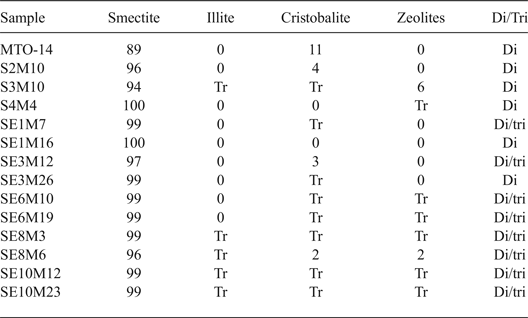
Detailed characterization of the samples was reported by Pérez del Villar et al. (Reference Pérez del Villar, Delgado, Reyes, Pelayo, Fernández-Soler, Cózar, Tsige and Quejido2005), Pelayo et al. (Reference Pelayo, García-Romero, Labajo and Pérez del Villar2011), Pelayo (Reference Pelayo2014) and Pelayo et al. (Reference Pelayo, García-Romero, Labajo and Pérez del Villar2016). The fine fraction (<2 µm) of bentonite samples from the White Tuffs formation, located both close to and away from the volcanic dome, consists mainly of dioctahedral smectite with minor illite, zeolites and cristobalite. The coexistence of dioctahedral and trioctahedral or intermediate di-trioctahedral smectite has been observed in some proximal samples (Table 2).
Table 2. Mineralogical composition (%) of the < 2 μm fraction from distal and proximal samples, characterised by XRD.
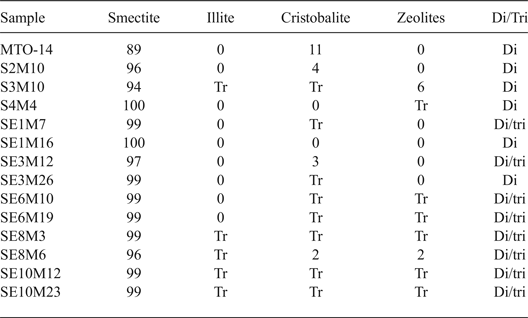
Tr: traces; Di/Tri: existence of dioctahedral and trioctahedral smectite.
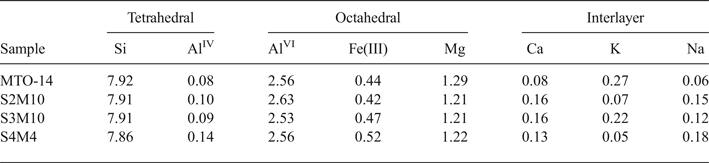
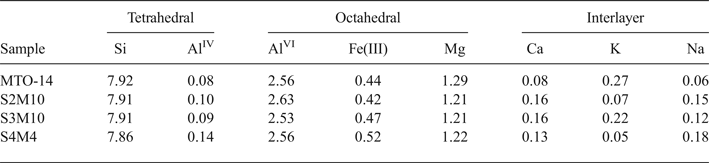
The structural formulae of smectites from distal samples (Table 3) correspond to Al-rich montmorillonites because of the prevalence of octahedral charge and the octahedral occupancy ranging from 4.21 to 4.33 cations per unit cell (p.u.c.).
Table 3. Structural formulae of smectites (calculated from TEM and EDX data) from distal samples (numbers of cations on the basis of O20 (OH)4).
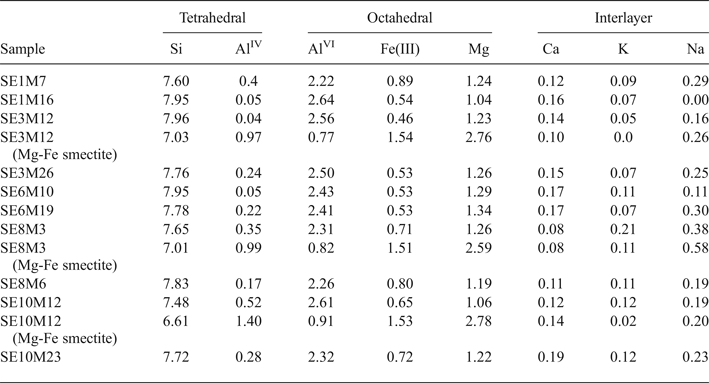
The smectites from proximal samples (Table 4) have a broader compositional variability. Thus, two groups were differentiated on the basis of their chemical composition: (1) Al-rich smectites, which are the more abundant in all the samples; and (2) Mg-Fe-rich smectites. The Al-rich smectites have mainly octahedral charge, except for samples SE1M7 and SE10M12 with a prevalence of tetrahedral charge. Furthermore, the number of Fe(III) cations is generally larger than that shown by distal smectites, with values > 0.6 p.u.c. in some samples, classifying them as Fe-rich montmorillonites or beidellites (Brigatti, Reference Brigatti1983). In summary, five proximal samples (SE1M16, SE3M12, SE3M26, SE6M10 and SE6M19) are Al-montmorillonites, three (SE8M3, SE8M6 and SE10M23) are Fe-rich montmorillonites and two (SE1M7 and SE10M12) are Fe-rich beidellites.
Table 4. Structural formulae of smectites (calculated from TEM and EDX data) from proximal samples (numbers of cations on the basis of O20 (OH)4).
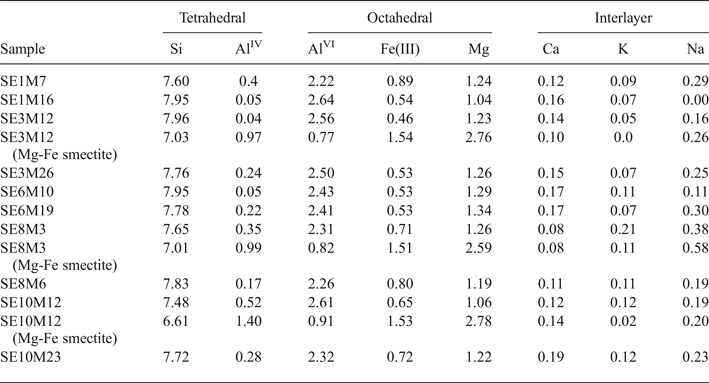
The structural formulae of smectites with high Fe and Mg contents present in three representative proximal samples (SE3M12, SE8M3 and SE10M12) are listed in Table 4. They show a prevalence of tetrahedral charge and octahedral occupancy ranging from 4.91 to 5.22 cations p.u.c.. Mg is the main octahedral cation, followed by Fe(III) and Al. Therefore, these smectites may be classified as intermediates between beidellite and Fe-rich saponite.
Methodology
A Fourier transform infrared (FTIR) spectroscopic study was carried out using a Nicolet 6700 FTIR spectrometer equipped with a DTGS detector and a KBr beam splitter, at a resolution of 4 cm−1, in transmission mode. 1 mg of sample was mixed with 200 mg of KBr to obtain pellets after pressing (10 tons max. load). The spectra per pellet sample were collected before and after heating at 110°C for 24 h in order to remove most of the adsorbed water. Analyses were carried out at room temperature in the Pore Water Chemistry Laboratory of the Environment Department, CIEMAT. The spectra were recorded in the mid-IR region (MIR), in the range 4000 to 400 cm−1. The OH-stretching region between 3800 and 3200 cm−1 and the OH-bending region between 1000 and 750 cm−1 were studied in detail.
Mössbauer spectra were recorded at room temperature (300 K) in transmission mode, using a constant acceleration spectrometer and a 57Co (Rh) source. An effective absorber thickness of approximately 5–10 mg Fe/cm2 was used in all cases. The velocity scale was calibrated using a 12 µm thick Fe foil and the isomer shifts were referred to the centroid of the spectrum of α-Fe at room temperature. With the aim of identifying the presence of Fe-oxyhydroxides, spectra were also recorded at 16 K for two samples using a closed-cycle He-cooled cryostat. The widths and areas of the two lines of a particular quadrupole doublet were assumed to be equal and those of the sextets had their areas held to a 3:2:1:1:2:3 ratio.
RESULTS AND DISCUSSION
Infrared spectroscopy
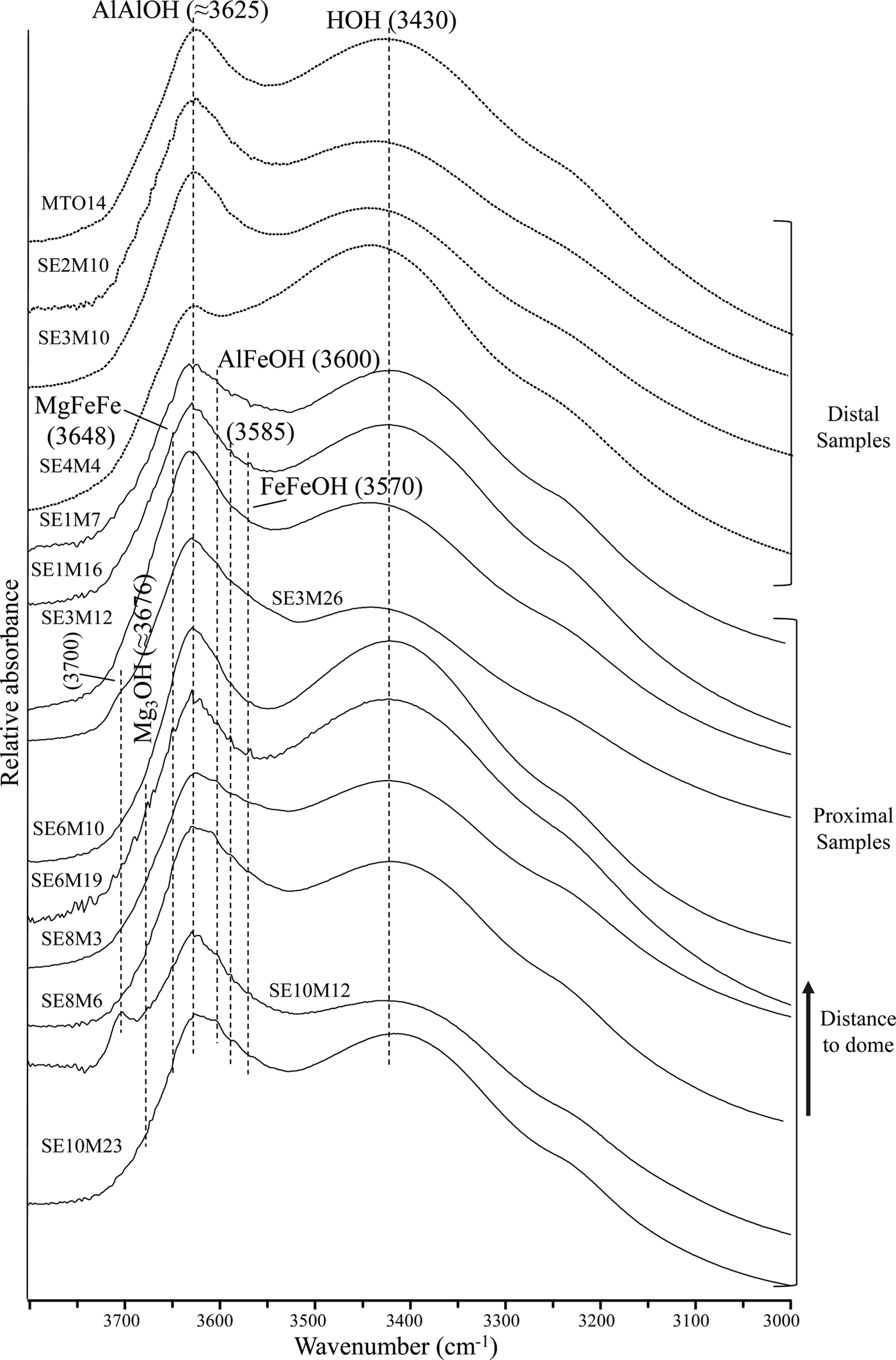
The FTIR spectra of the distal and proximal samples are shown in Fig. 2. The spectra show two main absorption bands in the OH-stretching region: one near 3625 cm−1, which is attributed to the AlAlOH vibration, typical of dioctahedral Al-rich smectites (Farmer, Reference Farmer1974); and a broader band at 3430 cm−1, assigned to the OH-stretching vibration due to molecular water absorption (Zviagina et al., Reference Zviagina, McCarty, Środon and Drits2004).
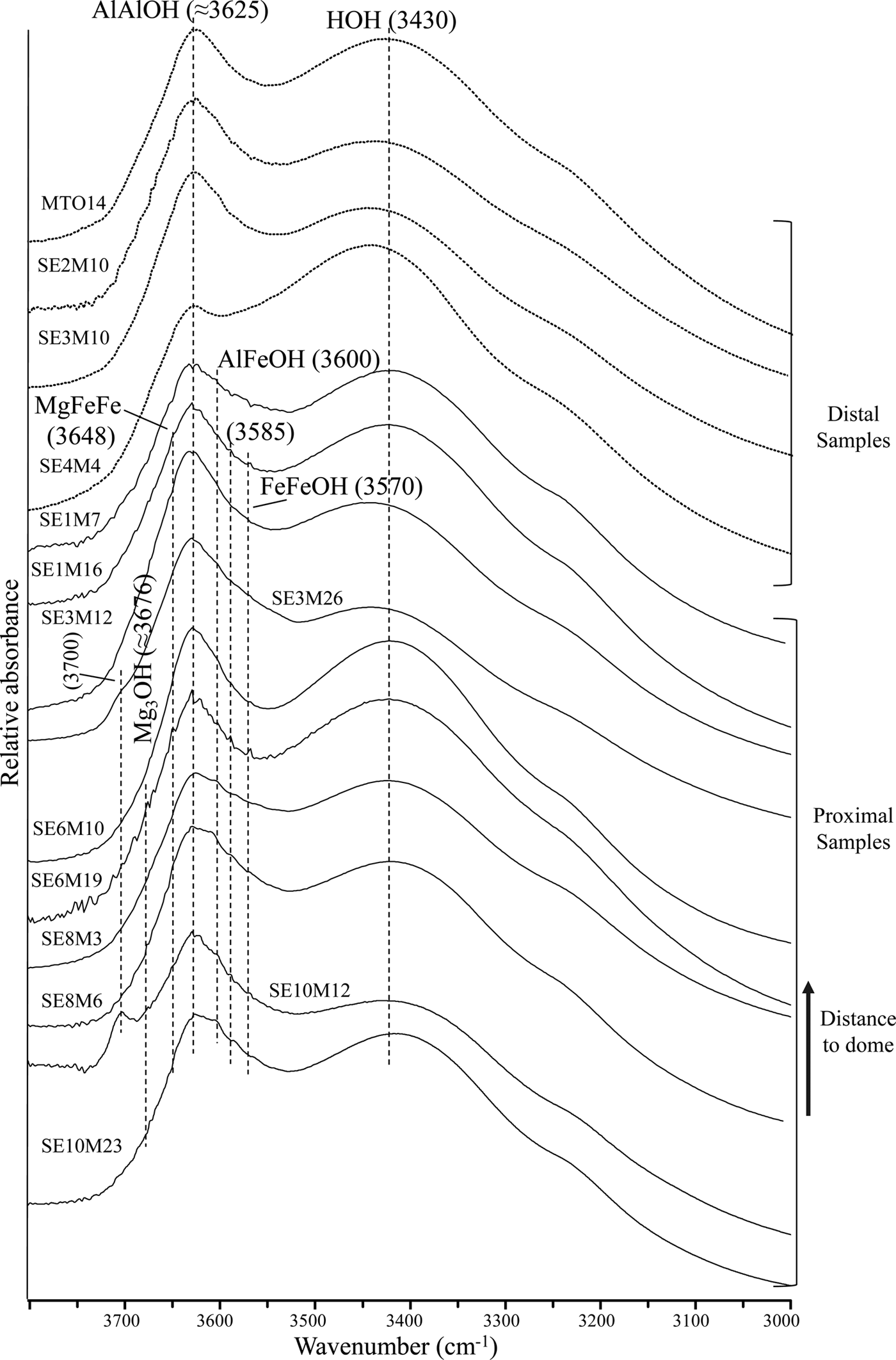
Fig. 2. FTIR spectra of distal and proximal samples in the OH-stretching region.
Furthermore, two shoulders near 3600 cm−1 and 3570 cm−1 are observed in most of the proximal samples. These bands correspond to AlFeOH and FeFeOH vibrations, respectively, of Fe-rich smectites (Bishop et al., Reference Bishop, Murad, Madejová, Komadel, Wagner and Scheinost1999; Komadel et al., Reference Komadel, Madejová and Stucki1999; Madejová et al., Reference Madejová, Bujdák, Petit and Komadel2000; Petit et al., Reference Petit, Caillaud, Righi, Madejová, Elsass and Köster2002). In addition, two weak shoulders near 3676 cm−1 and 3648 cm−1 are noted mainly in the proximal samples SE6M19 and SE10M12. The first shoulder is attributed to Mg3OH stretching vibrations of saponite (Farmer, Reference Farmer1974; Van der Marel & Beutelspacher, Reference Van der Marel and Beutelspacher1976; Parra et al., Reference Parra, Delmont, Ferragne, Latouche, Pons and Puechmaille1985), while the second may correspond to MgFe2OH stretching modes of Fe-rich saponites. Iron substitution in saponite shifts the Mg3OH stretching band towards a lower frequency (Wilkins & Ito, Reference Wilkins and Ito1967; Russell & Fraser, Reference Russell, Fraser and Wilson1994; Cuadros et al., Reference Cuadros, Dekov and Fiore2008). The existence of such bands is consistent with the presence of smectite with intermediate composition between beidellite and Fe-rich saponite in the proximal samples, in accordance with the structural formula calculations from TEM + EDX data (Table 3) (Pelayo, Reference Pelayo2014; Pelayo et al., Reference Pelayo, García-Romero, Labajo and Pérez del Villar2016).
Additionally, the band around 3700 cm−1 in the spectra from proximal samples SE3M26 and SE10M12 may be attributed to Mg3OH vibrations in dehydrated saponites containing interlayer Na or K (Farmer, Reference Farmer1974), chlorite/smectite mixed-layers (Van del Marel & Beutelspacher, Reference Van der Marel and Beutelspacher1976; Bergaya et al., Reference Bergaya, Brigatti and Fripiat1985) or to outer AlAlOH vibrations in kaolinite (Farmer, Reference Farmer1998; Madejová, Reference Madejová2003). As neither kaolinite nor chlorite/smectite mixed-layers was identified by XRD, the band at ~ 3700 cm−1 might be due to the presence of smectite with intermediate composition between beidellite and Fe-rich saponite, identified in sample SE10M12 (Pelayo, Reference Pelayo2014).
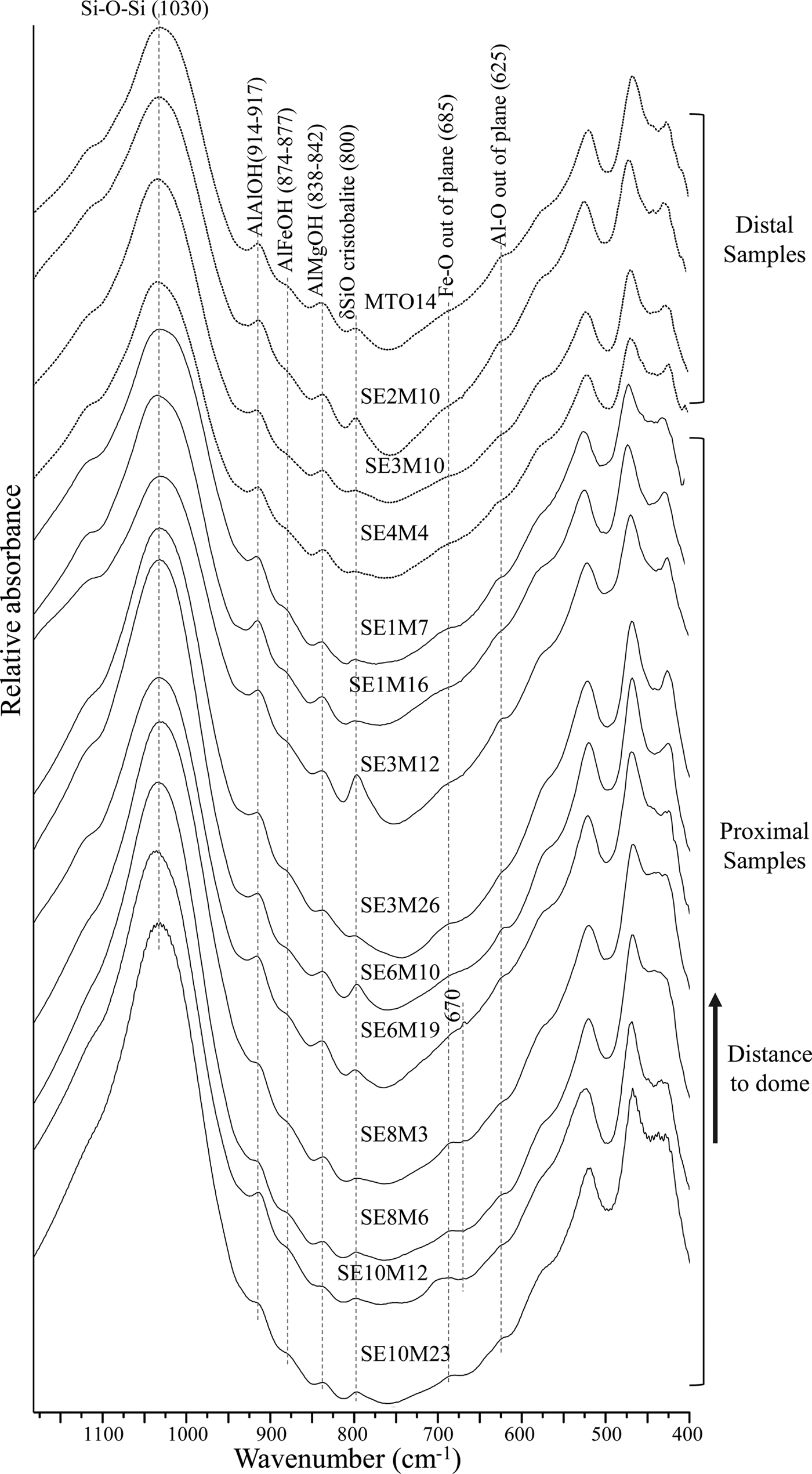
In the region between 1050 and 1000 cm−1 (Fig. 3), a strong band appears at 1030 cm−1, characteristic of Si-O vibrations of dioctahedral smectites (Goodman, Reference Goodman, Russell, Fraser and Woodhams1976).

Fig. 3. FTIR spectra of distal and proximal samples in the OH-bending region.
In the OH-bending region (Fig. 3), the three bands at 914-917 cm−1, 874-877 cm−1 and 838-842 cm−1, corresponding to AlAlOH, AlFeOH and AlMgOH vibrations of smectites (Farmer, Reference Farmer1974; Goodman et al., Reference Goodman, Russell, Fraser and Woodhams1976; Russell & Fraser, Reference Russell, Fraser and Wilson1994; Vantelon et al., Reference Vantelon, Pelletier, Michot, Barres and Thomas2001), are observed in all spectra. The AlFeOH vibration band is well defined in most proximal samples, and very weak in distal samples (Fig. 3). This indicates a higher Fe content in smectites from the proximal samples compared to their counterparts from distal samples, in agreement with the structural formulae (Table 3).
Furthermore, a band near 685 cm−1 appears in most of the proximal samples, which may correspond to out-of-plane OH-bending vibrations in Fe-rich smectites when the structure is disrupted by (1) tetrahedral substitution of Fe3+ for Si and/or octahedral cation substitution, or (2) by variations in hydroxyl position due to missing OH groups (Bishop et al., Reference Bishop, Madejová, Komadel and Froeschl2002). This band may be the out-of-plane bending vibrations predicted by Farmer (Reference Farmer1974) and may represent small trioctahedral domains within the dioctahedral smectite.
A weak shoulder around 670 cm−1 is observed in samples SE6M19 and SE10M12 which is assigned to Mg3OH vibrations in trioctahedral smectites (Farmer Reference Farmer1974). This fact, coupled with the presence of two shoulders near 3676 cm−1 and 3648 cm−1, seems to confirm the existence of saponite in the samples.
Additionally, some samples have a band at 625 cm−1 which corresponds to Al–O out-of-plane bending vibrations of Al-rich smectites with structural disorder due to tetrahedral and/or octahedral substitution (Bishop et al., Reference Bishop, Madejová, Komadel and Froeschl2002). Finally, the relatively strong band at 800 cm−1 observed in most samples is assigned to a SiO-bending vibration in cristobalite (Madejová & Komadel, Reference Madejová and Komadel2001), which was identified by XRD in the fine fractions of the samples (Table 2) (Pelayo, Reference Pelayo2014).
Mössbauer spectroscopy
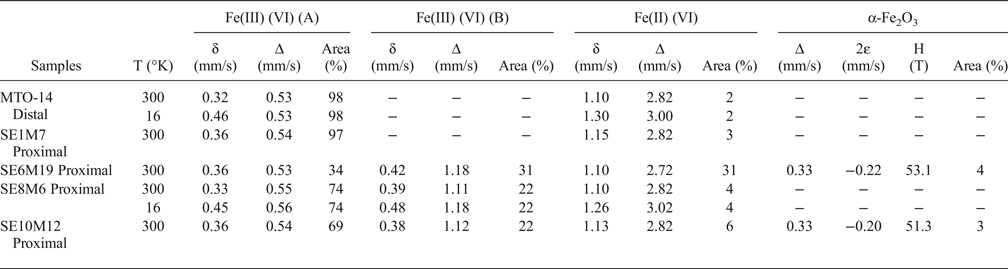
The Mössbauer spectra of the five samples studied are presented in Fig. 4. The Mössbauer parameters, the assignment to different Fe-species and their relative concentrations are listed in Table 5.

Fig. 4. Mössbauer spectra of the studied samples: (a) MTO-14 (distal); (b) SE1M7 (proximal); (c) SE6M19 (proximal); (d) SE8M6 (proximal); (e) SE10M12 (proximal); RT: room temperature (298 K).
Table 5. Mössbauer parameters obtained from the fit of the spectra recorded from the samples studied (mm/s), assignment to different Fe-species and their relative concentrations.
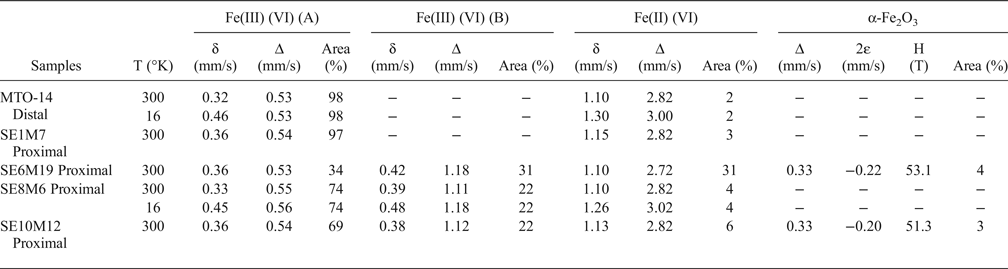
δ: Isomer shift; Δ: quadrupole splitting (in doublets); 2ε: quadrupole shift (in sextets); H: hyperfine magnetic field; (VI): octahedral coordination.
The Mössbauer spectrum of the distal sample MTO-14 recorded at 16 K is almost identical to that obtained at room temperature. Both spectra are dominated by an intense paramagnetic doublet (98% of the total spectral area) having Mössbauer parameters (δ = 0.32 mm/s; Δ = 0.53 mm/s) characteristic of Fe(III) in octahedral coordination (Bancroft et al., Reference Bancroft1973). In addition, both spectra show an additional small doublet with parameters (δ = 1.10 mm/s and Δ = 2.82 mm/s) characteristic of Fe(II) in octahedral coordination (Bancroft et al., Reference Bancroft1973). The absence of magnetic sextets at 16 K precludes the existence of Fe(III) oxides. Therefore, all the Fe(III) ions of the sample belong to the crystal structure of smectite.
The RT Mössbauer spectrum of the proximal sample SE1M7 is similar to that of the distal sample MTO-14, characterized by an intense paramagnetic doublet typical of Fe(III) in octahedral coordination, corresponding to 97% of the total Fe species and a small doublet, with parameters (δ = 1.15 mm/s; Δ = 2.82 mm/s), characteristic of Fe(II). The spectra of samples SE6M19, SE8M6 and SE10M12 have two doublets characteristic of octahedral Fe(III) with different quadrupole splitting value (∆) (Table 5). One doublet has a quadrupole splitting value of approximately 0.55 mm/s, whilst the second has a quadrupole splitting value ranging between 1.11 and 1.18 mm/s. The much larger quadrupole splitting value indicates a considerably more distorted octahedral environment for these Fe(III) octahedral cations (Maddock, Reference Maddock, Berry and Vaughan1985).
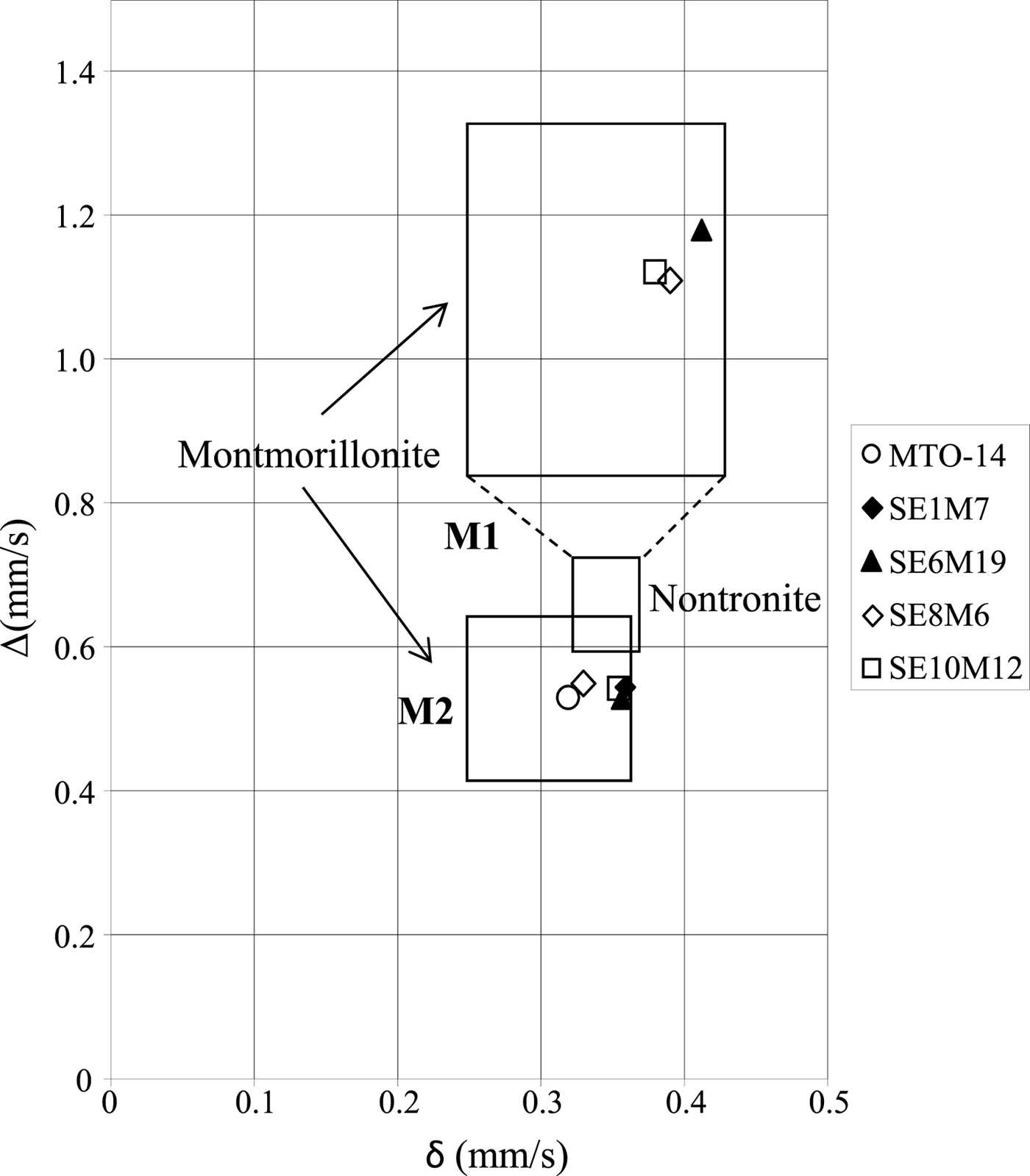
Other authors (Coey, Reference Coey1980; Heller-Kallai & Rozenson, Reference Heller-Kallai and Rozenson1981; De Grave et al., Reference De Grave, Vandenbruwaene and Elewaut1985) relate the two doublets with different quadrupole splitting values to Fe(III) ions in cis- and trans-octahedral sites. According to the diagram proposed by Coey (Reference Coey1980), the lower quadrupole splitting value is situated in the field of Fe(III) in cis positions (M2), while the higher value is located in the field of Fe(III) in trans positions (M1) (Fig. 5). Therefore, samples MTO-14 and SE1M7 with only one doublet might have Fe(III) in cis octahedral positions (M2). However, Cardile & Johnston (Reference Cardile and Johnston1986) suggested that Mössbauer doublets should not be assigned to cis- and trans-sites in the unit cell of 2:1 silicates, and this was further supported by independent studies which suggested that the observed differences of the quadrupole splitting are caused by a distortion of the Fe(III)-octahedron due to the different local structural and chemical environments (Dainyak & Drits, Reference Dainyak and Drits1987; Dainyak et al., Reference Dainyak, Drits and Heifits1992). Furthermore, the substitution of Al by Fe(III) causes distortion of the octahedron due to the larger ionic radius of Fe(III) (Rozalén, Reference Rozalén2004), which suggests that in the proximal samples a significant substitution of Al by Fe(III) in octahedral sheets took place. This fact is in agreement with the existence of the FTIR band at 685 cm−1, which indicates significant octahedral cation substitution.
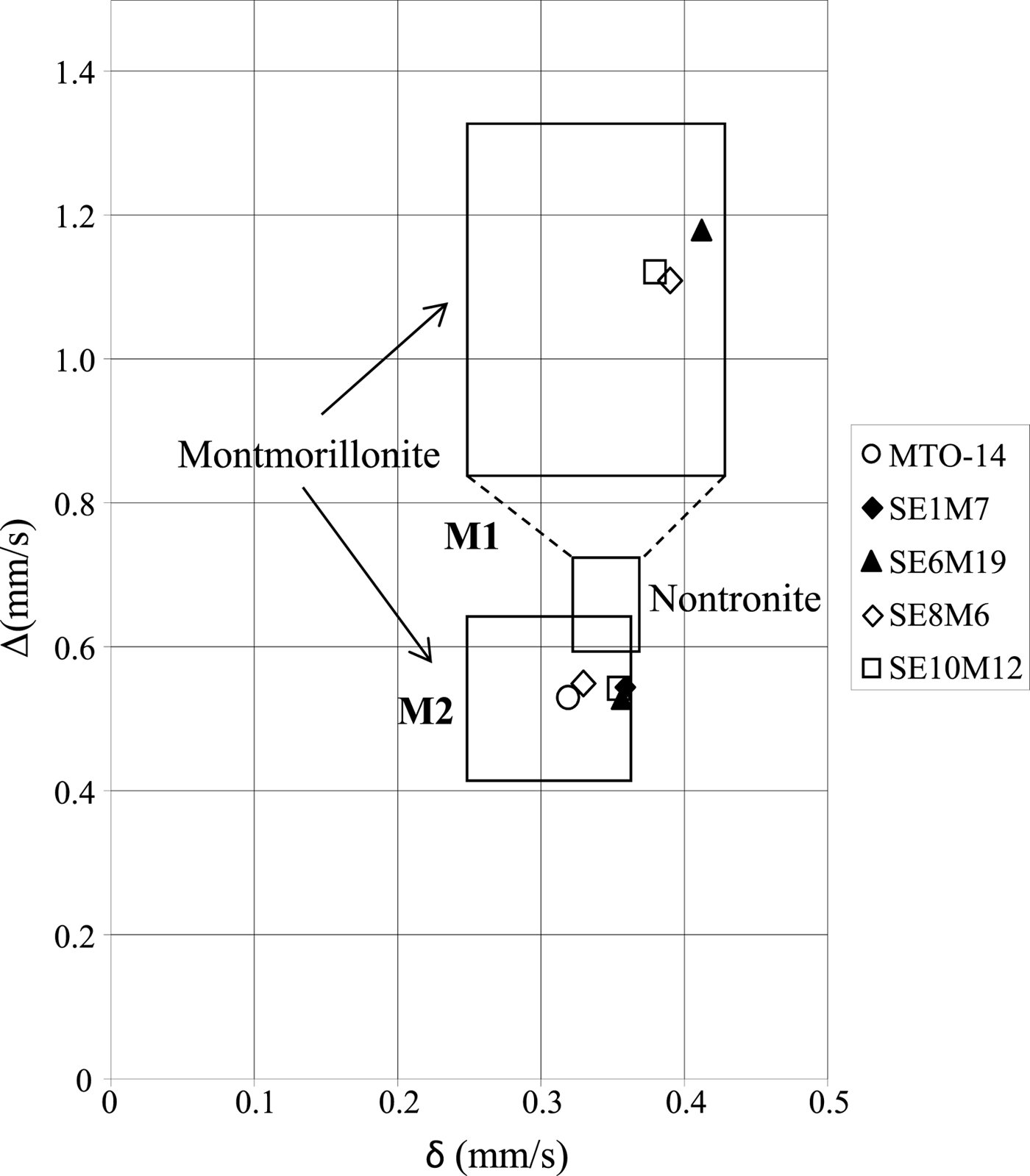
Fig. 5. Isomer shift (δ) vs. quadrupole splitting value (Δ) diagram proposed by Coey (Reference Coey1980). The M1 field corresponds to Fe (III) in octahedral trans positions, whilst the M2 field corresponds to Fe (III) in octahedral cis positions. The symbols correspond to the values of the samples studied in this work.
Additionally, the spectra from proximal samples also show a clear Fe(II) contribution that appears to be more intense than in the sample located away from the dome (MTO-14). This Fe(II) contribution is especially important in sample SE6M19, as it comprises ~30% of the overall Fe species of the sample. Furthermore, the spectrum of SE6M19 is very similar to that obtained by Beaufort & Meunier (Reference Beaufort and Meunier1994) in a saponite sample from clay deposits in fractures of a metamorphic basement, suggesting that sample SE6M19 might be composed of a mixture of montmorillonite and Fe-rich saponite, in agreement with the FTIR spectra.
Finally, only the proximal samples SE6M19 and SE10M12 showed the presence of a magnetically ordered phase. The corresponding sextet, accounting for 3–4% of the total Fe species, has hyperfine parameters, (δ = 0.33 mm/s; 2ε ≈ −0.21 mm/s; H ≈ 52 T) consistent with hematite (α-Fe2O3) (Dyar et al., Reference Dyar, Agresti, Schaefer, Grant and Sklute2006). These results indicate that the majority of the Fe(III) ions are in the crystal structure of smectites substituting for octahedral Al.
The structural formulae calculated from TEM and EDX analyses are in good agreement with the FTIR and Mössbauer results, confirming that the Al-smectites from the Morrón de Mateo bentonite deposit were transformed into Fe-rich smectites, mainly because of the volcanic intrusion which raised the temperature and supplied solutions. These solutions were initially enriched in Fe(II), transforming original Al-montmorillonites into Fe-rich montmorillonites and beidellites. These smectites would transform into Fe-rich smectites with an intermediate composition between beidellite and saponite in those areas with intense water–rock interaction. Finally, Fe(II) was oxidized to Fe(III) when bentonites were exposed to more oxidizing conditions close to the surface.
CONCLUSIONS
The IR spectra of the fine fraction of bentonites from the Morrón de Mateo deposit indicate that smectites from proximal samples have greater Fe-contents than their counterparts from samples further away from the dome. These last samples show OH-stretching and bending bands typical of Al-rich montmorillonite, while spectra from proximal samples show bands characteristic of both Al-rich smectites and Fe-rich smectites. Furthermore, most spectra of the proximal samples contain typical Fe-rich saponite bands. The IR data are in agreement with the structural formulae calculated in previous studies, indicating the presence of Fe-rich montmorillonite and smectite with intermediate composition between beidellite and Fe-rich saponite.
The Mössbauer data confirm that the Fe present in the fine fraction of bentonites is mainly located in the smectite structures, mainly as octahedral Fe(III). In two samples ~4% of total Fe is bound in hematite. Octahedral Fe(II) was identified as a minor component, except in one proximal sample where Fe(II) comprises 31% of the overall Fe species. This fact, and the characteristic FTIR bands, confirm the presence of Fe-rich saponite in this proximal sample.
Additionally, the Mössbauer spectra of proximal smectites contain two doublets characteristic of octahedral Fe(III). The much larger quadrupole splitting of one of these doublets indicates a much more distorted octahedral environment for this Fe(III) species, suggesting significant substitution of Al by Fe(III) in proximal samples.
The results confirm that alteration of smectites occurred in relation to the volcanic intrusion, which involved increase of temperature and supply of Fe-rich solutions responsible for the transformation of Al-montmorillonites into Fe-rich smectites.
ACKNOWLEDGMENTS
Financial support for this work was provided by ENRESA/CIEMAT and MINECO (Spain) through BARRA II (Contract number 774319) and MAT2015-64110-C2-1-P projects, respectively. The authors thank D. Tejela of Clariant Iberica Production, S.A. for providing the drill-core samples, and R. Saldaña for sample preparation. The authors also thank Martin Pentrák from the University of Illinois (USA) for the corrections, comments and suggestions that improved this work.