


Discussion and debate continues as to the congenital cardiac malformation currently warranting the dubious distinction as representing the worst disease. At one time, Roberts et al.1 considered hearts with aortic atresia as constituting the worst disease, at least in terms of survival. The emergence of strategies for functionally univentricular palliation, along with transplantation, for these patients, with ever improving outcomes, has markedly altered this perception.2 In a recent review,3 we discussed the continuing disappointing outcomes for patients with isomerism of the right atrial appendages, certainly justifying the nomination of this lesion as one of the current worst forms of disease. Few would deny, however, that the subset of patients with pulmonary atresia and intact ventricular septum with florid tricuspid regurgitation, the complication producing the so-called “wall-to-wall” heart (Figs 1 and 2), also have a very grim prognosis.4–6 The outcomes for the patients making up this subset, however, have received scant attention in comparison to that devoted to the more commonly encountered patients with hypoplastic ventricular cavities, severe right ventricular hypertension, and ventriculo-coronary arterial connections, another fascinating combination that received our recent attention.7 It is our intention to correct the neglect for those with dilated right ventricles in the present review.
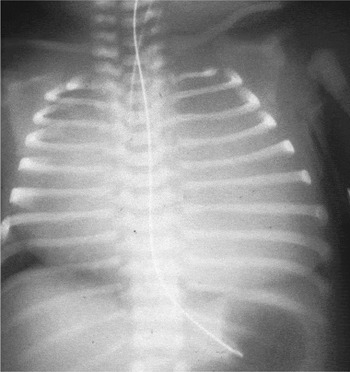
Figure 1. The chest radiograph, taken at post mortem, shows a wall-to-wall heart.

Figure 2. On opening the chest at autopsy of the patient shown in Figure 1, it can be seen that the heart (blue arrow) fills the majority of the thorax (yellow arrow), with the cardiomegaly due to dilation of the right atrium and right ventricle.
Historical Comment
As indicated above, we recently reviewed the findings in those patients with pulmonary atresia and intact ventricular septum complicated by ventriculo-coronary arterial connections, emphasizing their influence on surgical outcomes.7 In that review, we discussed the earliest attempt to classify hearts with pulmonary atresia and intact ventricular septum. This was undertaken by Greenwold et al., and published first as an abstract,8 and then expanded by Davignon et al.9 in a full manuscript, both publications emanating from the Mayo Clinic. While acknowledging the morphologic heterogeneity, these authors had suggested that the patients could be stratified into those with small as opposed to normal or large right ventricles. Fewer patients were placed into the group with normal or large ventricles, but the authors commented that, in their specimens justifying inclusion in this group, the lumen of the right ventricle was of normal size, albeit that mural thickness was increased.9 It is plain from this comment, therefore, that the group from Mayo clinic was not including those with dilated ventricles in their subset with “normal or large ventricles”. This simple classification remained popular through the 1970s, with Bharati et al.10 commenting that, while much attention had been devoted to the clinical and pathologic examination of patients with small right ventricles, much less attention had been given to those with the larger ventricles. It was this latter group that they considered to be potentially more amenable to surgery. The hearts reviewed by Bharati et al., 10 however, seem to have been those with dilated ventricles. As we will show, rather than being more amenable to surgery, these patients, characterized during life by their “wall-to-wall” hearts, still have a remarkably poor prognosis.
Incidence
Pulmonary atresia in the setting of an intact ventricular septum is not a particularly common malformation, having a prevalence of only 4.5 per 100,000 live-births. Those within this subset having massive tricuspid regurgitation, the hallmark of those with so-called “wall-to-wall” hearts, account for up to one-sixth of the overall group.7 The wall-to-wall heart is typically the consequence either of Ebstein's malformation,11, 12 or dysplasia of the normally attached leaflets of the tricuspid valve. While there has been some interest in the relationship between babies born with Ebstein's malformation as seen in isolation and maternal ingestion of lithium,13–15 such an association, as far as we know, has not been implicated for Ebstein's malformation as seen in pulmonary atresia with an intact ventricular septum. In the Baltimore-Washington Infant study, Ebstein's malformation was reported to have a prevalence of 5.2 per 100,000 live-births, only slightly higher than that found for all the infants with pulmonary atresia and intact ventricular septum.14 Hoffman et al., in contrast, suggest that Ebstein's malformation is seen only half as frequently in liveborn infants as pulmonary atresia and intact ventricular septum.16 The precise incidence of Ebstein's malformation as seen in the setting of an intact ventricular septum, therefore, has still to be established. And to date, as far as we are aware, no attempt has been made to calculate the incidence of those neonates born with dysplastic tricuspid valves in the absence of Ebstein's malformation.
Segmental and sequential analysis
The pulmonary valve is typically imperforate in those patients with pulmonary atresia and intact ventricular septum who exhibit the “wall-to-wall” malformation, although in some instances the atresia is functional rather than anatomic.12 The hearts themselves are usually left-sided, showing usual atrial arrangement, concordant atrioventricular and ventriculoarterial connections, and a left-sided aortic arch.7 There is a comparable lesion in the setting of aortic atresia with discordant atrioventricular and ventriculo-arterial connections.17–20 Patients with this combination can also exhibit the “wall-to-wall” arrangement, the morphologically right ventricle again being massively dilated due to massive regurgitation across the systemic morphologically tricuspid valve, and with either anatomic or functional aortic atresia.21–28 It is those with concordant atrioventricular and ventirculo-arterial connections and pulmonary atresia, however, on which we concentrate in this review. Such patients typically have confluent pulmonary arteries, fed by a left-sided arterial duct. The variations in pulmonary arterial anatomy, such as non-confluence, bilateral arterial ducts, and so on, that have been catalogued in patients with the hypertensive form of pulmonary atresia and intact ventricular septum,7, 29 are rarely, if ever, encountered in the subset with massive cardiomegaly due to the “wall-to-wall” arrangement.10, 15, 29, 30
Morphological substrates for the wall-to-wall heart
As already emphasized, the hearts to be discussed in this review differ significantly from those described by Davignon et al. as having normal or enlarged right ventricles.9 The essence of the wall-to-wall heart is its sheer size. The grossly enlarged heart, particularly the voluminous right atrium and enlarged right ventricle (Fig. 3), during the latter part of fetal development, occupies much of the volume within the bony thorax that should be available for the lungs. Despite the atrial enlargement, the caval veins and coronary sinus connect normally with the right atrium, and there is typically a true deficiency of the oval fossa (Fig. 3c). The tricuspid valve, however, is markedly dilated when normalized relative to body surface area. Nowadays, this measurement is described relative to normal values derived from the data of Rowlatt, Rimoldi, and Lev,31 thus giving the so-called Z-value.5 The more negative the Z-value, the smaller, and thus the more obstructive, is the orifice of the tricuspid valve. Such negative Z-values correlate in highly significant fashion both with the size of the right ventricular cavity and for the presence of ventriculo-coronary arterial connections.5 The larger Z-values, as may be expected, correspond with larger diameters, of the tricuspid valve, and hence more severe tricuspid regurgitation.5

Figure 3. As in the patient shown in Figure 2, this view of the opened thorax (a) in a patient with wall-to-wall heart shows that the heart fills the larger part of the thoracic cavity, squeezing out the lungs. The right atrium (b) is grossly dilated, and transillumination (c) shows the thinness of its walls.
When the tricuspid orifice is dilated because of co-existing Ebstein's malformation,10, 14, 15, 29, 30, 32–36 then as in the classical variants of the malformation, the leaflets attempt to form a competent valvar mechanism at the junction between the atrialized and functional parts of the right ventricle, rather than at the atrioventricular junction (Figs 3c and 4). The leaflets themselves, however, are grossly dysplastic, with wart-like and nodular features (Fig. 4). Wall-to-wall hearts (Fig. 5) can also be found when the grossly dysplastic leaflets are normally attached at the atrioventricular junction (Fig. 6). When the leaflets are deformed by Ebstein's malformation, the sail-like antero-superior leaflet typically retains its normal attachment at the atrioventricular junction (Fig. 4). Its leading edge, nonetheless, may be inserted in linear fashion in such a way as to obstruct the normal outflow beneath its leading edge to the right ventricular outflow tract.32, 37 Thus, while the most recognized functional correlate of the tricuspid valve deformed by Ebstein's malformation is that of regurgitation, the abnormal valves can also exhibit some degree of functional stenosis. Indeed, in rare circumstances, the valve deformed by Ebstein's malformation can be imperforate, producing a variant of tricuspid atresia.32, 37–40 The hearts seen with tricuspid stenosis or atresia, however, do not typically show exorbitant cardiac enlargement, and hence do not fall within the category of “wall-to-wall” heart.

Figure 4. The valve from the heart shown in Figure 3 exhibits Ebstein's malformation. The septal leaflet is no more than a warty excrescence formed on the septum well away from the atrioventricular junction (dotted line). The antero-superior leaflet has a normal attachment at the atrioventricular junction, and focal distal attachments, but is markedly dysplastic. The mural leaflet has failed to delaminate. Note the dilated atrialised portion of the right ventricle (RV).

Figure 5. Opening the thorax in this case again shows a wall-to-wall heart, due to dilation of the right atrium and ventricle.
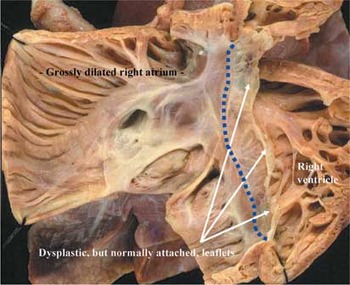
Figure 6. Opening the right atrioventricular junction of the heart shown in Figure 5 reveals that the leaflets of the tricuspid valve, although grossly dysplastic, are normally attached at the atrioventricular junction (dotted line).
When the hearts do show the “wall-to-wall” malformation, then the right ventricular myocardium always exhibits some degree of thinning (Fig. 7), unlike the situation in the enlarged hearts described by Davignon et al.,9 which possessed hypertrophied right ventricular walls. The thinning involves not only the atrialized portion of the right ventricle, as is typical for when the tricuspid valve itself is deformed by Ebstein's malformation,10, 14, 15, 29, 30, 32, 33 but also the remaining functional part of the morphologically right ventricle (Fig. 7). In this respect, some have correlated the thinning seen in the setting of the wall-to-wall heart with the arrangement seen in Uhl's anomaly of the right ventricle.41–43 This is inappropriate. The changes in the right ventricular wall itself are acquired due to the ventricular dilation. In Uhl's anomaly,44, 45 the changes are exclusively congenital, since the tricuspid and pulmonary valves themselves are normal.

Figure 7. In the heart shown in Figures 3 and 4, the myocardium is thinned throughout the dilated walls of the right ventricle.
In the “wall to-wall” heart, the pulmonary outflow tract is expected to be completely sealed and imperforate. In some patients, however, the atresia may be functional as opposed to anatomic, albeit that it is frequently difficult to make this distinction (see below).5 We were able to make this clinically important distinction more than 25 years ago using prostaglandin-enhanced retrograde aortography.15, 29 In recent years, the emergence of colour-flow Doppler interrogation has facilitated non-invasive distinction.46–49
The pulmonary trunk usually bifurcates normally into right and left pulmonary arteries, these arteries usually exhibiting some degree of hypoplasia, frequently severe. The compressed lungs appear underdeveloped and hypoplastic.29, 50, 51 Although appearing grossly hypoplastic, it does not follow that the substance of the lungs is itself abnormal. Thus, Tanaka et al.50 failed to find any significant alveolar underdevelopment or immaturity of the parenchyma, nor were they able to recognize abnormal degrees or extent of muscularization of the pulmonary arteries. Similar findings were reported by Lang et al.51 This knowledge is important for those seeking to manage and manipulate the flow of blood to the lungs in these patients. The changes are markedly different from those seen in patients with small right ventricles in the setting of pulmonary atresia with intact septum, in whom the pulmonary arteries are too small, too few in number, and have too thin a muscular coat.52–54 It is not surprising that pulmonary arterial and parenchymal hypoplasia has also been observed in those patients with aortic atresia, discordant atrioventricular and ventriculo-arterial connections, severe tricuspid regurgitation, and remarkable cardiac enlargement.27, 28
The pulmonary veins connect in a normal fashion to the left atrium.50, 51 When compared to the gigantic right atrium, the left atrium appears small and under-filled. Mild dysplasia of the mitral valve, with functional prolapse, has been recorded in the older patient with isolated Ebstein's malformation of the tricuspid valve.55 We have observed similar findings in those patients having Ebstein's malformation in the setting of pulmonary atresia, intact ventricular septum, and severe tricuspid regurgitation, albeit that typically the left ventricle, and aortic outflow tract, is grossly normal (Fig. 8). Left ventricular myocardial non-compaction has been reported in a few older patients with isolated Ebstein's malformation of the tricuspid valve,56 although this is uncommon. As far as we are aware, such noncompaction has not been observed in the left ventricles of the sev-erely symptomatic neonates with wall-to-wall heart, although it has certainly been seen in the hypertensive forms associated with ventriculo-coronary arterial connections.19, 29 It is perhaps not surprising that Ebstein's malformation, or dysplasia, of the tricuspid valve can coexist with pulmonary atresia and intact ventricular septum. It is now well established that anatomic pulmonary atresia due to an imperforate pulmonary valve develops relatively late in gestation, being a consequence of both a disturbed intracardiac anatomy promoting a lower than normal flow of blood through the right ventricle and pulmonary trunk, and disturbed fetal haemodynamics, with intrauterine progression of pulmonary valvar obstruction.57–63

Figure 8. The left ventricle from the heart shown in Figures 3 and 7 is basically normal.
Relatively few other lesions are encountered in the neonate that produce such important and remarkable cardiomegaly to qualify as a wall-to-wall heart.15, 19, 21–30 The majority of these other conditions are observed in patients with low pressures in the right ventricle, and right-to-left shunting at atrial level.15, 29, 30 When making the diagnosis, nonetheless, the application of fetal and perinatal echocardiographic techniques, or other modes of cardiac imaging, will readily identify the patients with discordant atrioventricular connections and functional or anatomic aortic atresia, and will similarly identify those neonates with normal sequential and segmental cardiac anatomy who exhibit an intrapericardial teratoma or other miscellaneous condition, such as an aneurysm of the vein of Galen.
Fetal recognition and outcome
As discussed above, the progression from critical pulmonary stenosis to atresia is now well recognized during fetal life.57–63 Others have studied the impact of florid tricuspid regurgitation on the ongoing gestation.64–69 It is hard to define the impact on the fetus, nonetheless, of associated pulmonary atresia, and to determine whether this reflects an anatomic or functional disturbance. Daubeney et al.70 have addressed the impact of fetal echocardiography on the incidence at birth, and postnatal outcome, of the entire cohort of patients with pulmonary atresia and intact ventricular septum. Thus, from 1991 to 1995, a register was made of all infants born with this lesion in the United Kingdom and Eire, and of all diagnoses of this condition made during fetal life. During this period, there were 183 live births, giving an incidence of 4.5 for each 100,000 live births. The incidence varied, with statistical significance, amongst the countries making up the United Kingdom, being 4.1 cases per 100,000 live births in England and Wales, 4.7 in Scotland, 9.6 in Northern Ireland, and 6.8 in Eire. Diagnosis had been made during fetal life in 86 cases, at a mean of 22.0 weeks of gestation, and in 53 cases, the pregnancy had been terminated. Of the remainder, there were 4 intrauterine deaths and 29 live births. Had there been no terminations of pregnancy, the incidence at birth would have been 5.6 per 100,000 births in England and Wales, 5.3 in Scotland, but would have remained unchanged in Northern Ireland and Eire, presuming no further spontaneous fetal deaths. Severe tricuspid regurgitation had been identified in 20 of the fetuses, and 9 of these were thought to have Ebstein's malformation of the tricuspid valve. There was no difference in outcome for the 29 live-born cases recognized during fetal life, and the remaining 154 live-born infants not diagnosed prenatally. A companion paper, published several years later,71 revealed that, amongst the 183 live-born infants, Ebstein's malformation was found in 18 patients, albeit not with “wall-to-wall” hearts.
At the Hospital for Sick Children, we made a diagnosis of pulmonary atresia with intact ventricular septum in 34 fetuses between the years 1999 and 2004. Our experience emphasizes the significance of anomalies of the tricuspid valve in these patients when diagnosed prenatally, since 7 of the fetuses had Ebstein's malformation (Fig. 9), and 4 had dysplastic tricuspid valves (Fig. 10). These anomalies resulted in severe tricuspid regurgitation in 9 fetuses, and moderate regurgitation in 2, with cardiomegaly and significant right atrial enlargement present in all these cases (Figs 9 and 10). Indeed, 5 fetuses already exhibited hydrops when first diagnosed. Only two of the fetuses, both diagnosed late in gestation, with Ebstein's malformation in one and tricuspid valvar dysplasia in the other, did not die during the fetal or neonatal periods, emphasizing the particularly poor outlook of fetuses with coexistent pulmonary atresia, be it structural or functional. In our first fetus who survived, with Ebstein's malformation, the pulmonary valve was noted to be imperforate at birth. This patient underwent successful balloon valvoplasty as a neonate. In the patient with tricuspid valvar dysplasia, the atresia proved to be functional (Fig. 11), and resolved completely, along with the obstruction at the right ventricular outlet, once the pulmonary arterial pressure had dropped after birth.

Figure 9. This fetal echocardiogram shows an enlarged heart due to Ebstein's malformation (a) producing severe tricuspid regurgitation (b).

Figure 10. The fetal echocardiogram (a) again shows gross dilation of the right atrium and ventricle, but this time due to a dysplastic tricuspid valve (compare with Figure 9). Doppler interrogation (b) reveals florid tricuspid regurgitation.

Figure 11. This echocardiogram is from the same fetus as shown in Figure 10. Doppler interrogation has demonstrated reversed flow in the arterial duct due to functional pulmonary atresia.
Functional versus anatomic atresia
When the tricuspid valve is grossly regurgitant, it is important to differentiate functional from organic or anatomic pulmonary atresia.15, 29, 30 During fetal life, however, it is often impossible to make this distinction in the absence of pulmonary regurgitation, since reversed flow is detectable in the arterial duct in both situations. In those patients with a patent outflow tract, but functional obstruction to the flow of blood to the lungs, time combined with medical manipulation of the pulmonary circulation after birth may eventually set the scene for antegrade flow to the lungs. As shown by our own second case diagnosed during fetal life, this can obviate in some the need for surgical intervention in the neonatal period. In this subset, time allows for the normal vascular remodeling of the pulmonary circulation, with improvement in its compliance.15, 19, 29, 30, 72–74 Furthermore, in the patients known to have functional obstruction, dilation of the pulmonary vascular bed with nitric oxide may prove beneficial.74–76 These considerations obviously have little relevance when the obstruction to flow is anatomic rather than functional. If the pulmonary valve is patent but severely incompetent, this added complication will only worsen the functional state of the grossly regurgitant tricuspid valve.15, 29 Administration of prosta-glandin to maintain ductal patency may also prove helpful, at least in the short-term, for some severely affected neonates with either Ebstein's malformation or tricuspid valvar dysplasia, be the obstruction at the ventriculo-arterial junction due to either anatomic or functional pulmonary atresia.
Survival
More than 15 years ago, we identified in Toronto the factors influencing survival of patients with pulmonary atresia and intact ventricular septum.4 Operative weight at first operation, the ratio of right to left ventricular pressures, and absence of ventriculo-coronary arterial connections, were identified as significant predictors of survival. For those with a low ratio of the right to the left ventricular pressure, this being a deleterious finding, this was usually the consequence of severe tricuspid regurgitation in the setting of a thinned right ventricular myocardium, usually reflecting the “wall-to-wall” heart seen with Ebstein's malformation, or tricuspid valvar dysplasia. This initial experience has now been extended by Dyamenahalli et al.,77 who analysed the 210 consecutive patients seen in Toronto with pulmonary atresia and intact ventricular septum from 1965 to 1998. Overall survival was 72% at age 1 month, 57% at 1 year, 48% at 5 years and 43% at 10 years. Only earlier date of birth, and the presence of Ebstein's malformation with prematurity, proved to be significant independent factors associated with decreased overall survival. After controlling for these 2 factors, no other factors significantly impacted on survival. Those patients with a low ratio of right to left ventricular pressures together with Ebstein's malformation, however, continued to have a poor outcome. Indeed, the patients with severe tricuspid regurgitation accounted for almost one-fifth of the entire cohort, and also made up one-twelfth of our entire cohort identified with Ebstein's malformation of the tricuspid valve. Predicted actuarial survival for any era would be enhanced if this particular group had been excluded from analysis.
Data provided from the Society of Congenital Heart Surgeons5 also proves interesting when exploring the outcomes of the patients with severe tricuspid regurgitation. In their study, a severely incompetent tricuspid valve was observed in just over one-quarter of the patients. They showed that increasing severity of tricuspid incompetence was correlated with poorer outcome, albeit that it is difficult to correlate the degree of tricuspid valvar regurgitation with specific morphology, such as Ebstein's malformation as opposed to dysplasia of the tricuspid valve. It can be deduced, nonetheless, that the severity of tricuspid regurgitation, and thus larger right ventricular cavity size, did not correlate with the presence of ventriculo-coronary arterial connections, nor with a right ventricular-dependent coronary circulation. The experience reported by the Congenital Heart Surgeons has recently been updated by Ashburn et al.6 They analysed 408 neonates with pulmonary atresia and intact ventricular septum, registered prospectively between 1988 and 1997 by 33 participating institutions. Overall survival was 77% at 1 month, 70% at 6 months, 60% at 5 years, and 58% at 15 years. Of the patients, 15 years after entry to the study, one-third had been converted to biventricular circulations, one-fifth had the Fontan circulation, one-twentieth had undergone a 1.5 ventricle repair, transplantation had been performed in 2%, but almost two-fifths had died before reaching definitive repair. The remaining 2% were alive without definitive repair. Again, they discovered that severe right ventricular enlargement and tricuspid regurgitation, reflecting Ebstein's malformation of the tricuspid valve or primary tricuspid valvar dysplasia, imposed a high risk of death and failure to achieve a biventricular repair. Interestingly, for those patients with a diminutive right ventricle, and ventriculo-coronary arterial connections, particularly with a right ventricular-dependent coronary circulation, introduction of functionally univentricular palliation has shown a striking trend for improval.78, 79 But no such trend for improvement has been observed for those patients with severe tricuspid regurgitation. Indeed, comment has been made on the reticence to discuss the patients with “wall-to-wall” hearts.80
Is it possible, therefore, to contemplate surgical salvage for the patients with pulmonary atresia, intact ventricular septum, very severe tricuspid regurgitation, and a thinned right ventricle? These patients are unequivocally less amenable to satisfactory surgical intervention than those with a smaller right ventricle and a disadvantaged coronary circulation due to the presence of ventriculo-coronary arterial communications.7 Some patients with very severe tricuspid regurgitation have survived subsequent to construction of a systemic-to-pulmonary shunt, or after surgical or catheter-based intervention on the atretic pulmonary valve, combined with some form of tricuspid valvoplasty and reduction or reconstruction of the grossly dilated chambers of the right heart.81, 82 The approach initially taken by Starnes and his coworkers,83 nonetheless, has proven efficacious in salvaging some of these babies. They83 opined that the critically-ill babies could best be treated by converting the regurgitant tricuspid valve to an atretic one, combining this with construction of a systemic-to-pulmonary arterial shunt, followed later by further functionally univentricular palliation. In their patients, the pulmonary outflow tract was anatomically patent, but there was functional pulmonary atresia. All 5 babies survived this new operation, with 2 proceeding to successful conversion to the Fontan circulation, and one receiving a successful cavopulmonary shunt. Since this report,83 a number of case reports, and small series, have confirmed the merits of the approach, some combining the initial surgery with reduction of the right atrial wall.84–88 Experience with the procedure is also included in the report from the Pediatric Cardiac Care Consortium,89 with 5 of the 462 patients with pulmonary atresia and intact ventricular septum enrolled by the participating members undergoing surgical closure of the tricuspid valve, with two deaths. There is some confusion, however, since outcomes have also been analysed for the 215 patients with Ebstein's malformation enrolled by the members of the same consortium.90 This analysis showed that the tricuspid valve had been closed in 6 patients, with 4 deaths. When we consider that pulmonary atresia, an intact ventricular septum, and severe tricuspid regurgitation, with its grossly disadvantaged right ventricle, comprises up to one-sixth of all patients with pulmonary atresia and intact ventricular septum, we must conclude that the innovative approach taken by Starnes and his colleagues has done relatively little to salvage the majority of such affected babies. Knott-Craig et al.91, 92 have reported some recent success with neonatal repair of Ebstein's malformation, including some patients with functional or anatomic pulmonary atresia, but the overall experience remains small.
We wonder, therefore, whether there is a role for fetal intervention93–95 in this particular group of patients? In the absence of severe pathology involving the right ventricle and the tricuspid valve pathology, fetal intervention has been already successfully accomplished in a few cases. The number of patients benefiting in this way is very small. Perhaps the answer rests in defining whether a perforate outflow tract confers any possible advantage to the immediate care of the neonate? The presence of organic pulmonary atresia does not allow for any medical manipulation of the pulmonary vascular bed. Yet, if intrauterine balloon dilation of a progressively stenotic outflow tract results in important pulmonary regurgitation, will that further aggravate an already egregious situation?96 The answers to these questions remain speculative. Perhaps more is to be gained from understanding those regulatory processes important to the formation of the “wall-to-wall” hearts. There is now a canine model of Ebstein's malformation as seen in the human, in which the gene responsible has been mapped to the canine chromosome 9.97 We might hope that such animal models will provide the window necessary to understand the cardiac dysmorphogenesis represented by hearts with pulmonary atresia, intact ventricular septum, and Ebstein's malformation or dysplasia of the tricuspid valve, acknowledging that currently this form of congenital cardiac lesion continues largely to frustrate surgical intervention.
Summary
From our own experience, combined with review of the literature, we conclude that the variant with a grossly regurgitant tricuspid valve accounts for up to one-sixth of all patients with pulmonary atresia and intact ventricular septum, and an even greater proportion when the lesion is diagnosed during fetal life. The basis for the severe tricuspid regurgitation is either Ebstein's malformation, or a severely dysplastic tricuspid valve with normal annular attachments. Because of the regurgitation, the right ventricle is disadvantaged overall by marked thinning of its wall. The patients themselves typically demonstrate remarkable cardiac enlargement, with compromised cardiopulmonary function. They continue to have a particularly poor prognosis, with little substantial improvement shown over time. Whilst some of these patients may benefit from the Starnes operation, a procedure converting the initial biventricular anatomic arrangement to a functionally univentricular pattern, the constellation of malformations must certainly be considered to constitute one of most current egregious forms of congenital cardiac disease.
Acknowledgements
We thank Ruth Taylor, of the Division of Cardiology for outstanding secretarial support.
Professor Anderson is supported by grants from the British Heart Foundation together with the Joseph Levy Foundation.
Research at the Institute of Child Health and Great Ormond Street Hospital for Children NHS Trust benefits from R&D funding received from the NHS Executive.









