Congenital long QT syndrome (LQTS) results in abnormal ventricular re-polarisation in patients with otherwise normal hearts. Jervell and Lange–Nielsen first described the disorder in 1957 when an association between congenital deafness, prolongation of the QT interval, and an increased risk of sudden death in a single family was observed.Reference Jervell and Lange-Nielsen 1 Shortly after this initial description, Romano and Ward also described a similar association but in patients not having deafness.Reference Ward 2 Of the cardiac channelopathies, LQTS is the most common, affecting an estimated 1 in 2000 people and accounting for an estimated 5000 deaths per year in the United States of America.Reference Schwartz, Stramba-Badiale and Crotti 3 , Reference Tester, Will and Haglund 4 Owing to the rate at which deaths occur during exercise and competitive athletics, it has become crucial for clinicians to understand risk stratification and to make informed decisions regarding the safety of sports participation with respect to LQTS.
Following the initial clinical descriptions of LQTS, there has been a great deal of investigation into the genetic aetiology and pathophysiology of these entities, with goals of improving screening tools and understanding associated risks. The diagnosis of LQTS in the modern era remains clinical and is based on family and personal history of symptoms and resting and exercise electrocardiograms. The Schwartz Score is often used to quantify the probability of congenital LQTS in any given patient.Reference Schwartz and Crotti 5 Today, the genetic basis for LQTS has been pinpointed for 75–80% of affected patients, with more than a dozen genes identified including three genes which account for 75–90% of gene-positive LQTS. Early investigators suggested that pre-symptomatic diagnosis and treatment of this life-threatening disorder may be possible.Reference Ackerman 6 Early identification, in addition to more effective therapeutic strategies, has altered the prognosis of LQTS. We have come to find that the risk of adverse events is dependent on the specific genotype and mutation a patient carries along with the patient’s age, sex, and degree of QTc interval prolongation.Reference Liu, Jons and Moss 7 Through this research, heart rhythm experts continue to revise their recommendations regarding sports eligibility.
Mechanisms of exercise as an arrhythmia trigger in LQTS
Although great strides have been made in the genetics underlying the various defects in cardiomyocyte ion channels and their associated proteins, the cellular basis for arrhythmia generation has been more elusive. Earlier studies on the cellular mechanisms underlying LQTS demonstrated that electrical heterogeneity within the ventricular myocardium in these patients acts as a substrate for torsade de pointes ventricular tachycardia. Furthermore, it was found that M cells, located deep in the ventricular myocardium of humans, have unique properties due to their pronounced IKs – slow rectifying potassium current (see below) – relative to that of other ventricular cardiomyocytes. This predominance of IKs creates a particular vulnerability to QT interval-prolonging agents, which leads to transmural heterogeneity of re-polarisation.Reference Antzelevitch and Shimizu 8 Transmural dispersion of refractoriness during re-polarisation within the myocardium is a set-up for unidirectional block within small areas of the myocardium, and this in turn is a substrate for micro-reentry and ultimately torsade de pointes. In order for arrhythmia to occur, however, there must be a trigger, which in the case of LQTS is early after depolarisations within the ventricular myocardium. This trigger results from inward calcium flow through L-type channels, which occurs more under the influence of increased sympathetic tone.Reference Keating and Sanguinetti 9 Therein lies the invocation of exercise as it pertains to relative risk in LQTS. Investigators have been able to explore the unique influence sympathetic tone has on each of the ion channels involved in LQTS. Dissecting these important genotype differences is critical to understanding risk as it pertains to exercise in LQTS.
LQT1
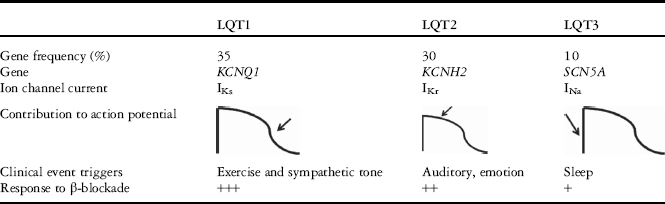
LQT1, the most common form of LQTS, is caused by a mutation in the KCNQ1, previously called KVLQT1, gene on chromosome 11 (Table 1). This gene encodes the α subunit of the Kv7.1 potassium channel, which is responsible for the slow delayed rectifying potassium current (IKs) predominant during phase 3 of ventricular myocyte re-polarisation. During the myocyte action potential, it is this current that stabilises the charge across the cell membrane and acts to return the cell to its baseline polarised state. The IKs in fact contributes very little to the action potential under basal conditions because of its slow activation compared with the rapid delayed rectifying current (IKr), which is affected in other forms of LQTS.Reference Jost, Viraq and Bitay 10 This channel in particular is crucial to the adaptive shortening of myocyte re-polarisation during sympathetic activation.Reference Tseng and Xu 11 When this channel is defective because of a loss-of-function mutation, as is the case with LQT1, re-polarisation fails to shorten, which is manifest by a prolonged QT interval. This has been confirmed to be the case in several studies, the largest of which demonstrated that out of 392 patients with LQTS 62% had exercise as a trigger.Reference Schwartz, Priori and Spazzolini 12 As would be expected, these patients exhibit QT interval prolongation during peak exercise, and these intervals remain prolonged for an extended amount of time during exercise recovery.Reference Aziz, Wieand and Ganley 13
Table 1 Common forms of long QT syndrome (LQTS).

LQT2
LQT2 is the second most common form of LQTS and is caused by mutations of the KCNH2 gene on chromosome 7 (Table 1). This encodes HERG, the α subunit of the rapid delayed rectifier potassium current (IKr), which along with the slow delayed rectifier current (IKs), is responsible for the delayed rectifier potassium current that determines phase 2 of ventricular cardiomyocyte re-polarisation. The predominant trigger for arrhythmias in these patients is auditory stimuli, which in one study occurred in 43% of LQT2 patients with cardiac events. The next most common circumstance for arrhythmia in these patients, however, is sleep without arousal.Reference Schwartz, Priori and Spazzolini 12 In contrast to patients with LQT1, these patients exhibit appropriate QT interval shortening during peak exercise, which is the normal adaptation seen in people without LQTS in order to allow for increased heart rates without a pro-arrhythmic substrate. This is also why these patients are at much lower risk for arrhythmia during exercise, although 13% of patients still have been reported to have events with activity.Reference Schwartz, Priori and Spazzolini 12
LQT3
LQT3 is the third most common form of LQTS and is caused by a gain-in-function mutation of the SCN5A gene on chromosome 3, which encodes the α subunit of the cardiac sodium channel (Table 1). This channel is responsible for phase 0 of the action potential, which is characterised by depolarisation of the cardiomyocyte via the inward flow of positively charged sodium. Owing to the gain-of function of this channel, there is re-opening during phase 2 of the action potential, and this effectively prolongs re-polarisation. Although this is still a set-up for arrhythmia, the sodium channel is less vulnerable to sympathetic tone. Patients with LQT3 tend to have arrhythmias triggered during sleep or rest.
Current status of risk stratification in LQTS
Sports eligibility is largely predicated on the basis of our clinical interpretation of risk, which has evolved into a more personalised equation since the discovery of LQTS. In 1998, when only four genes linked to LQTS had been discovered, Zareba et al were the first to demonstrate that distinct genotypes were associated with different level of risk for cardiac events. This marked the beginning of risk stratification based on genetic testing for this disorder and was a milestone for the recognition of the field of genotype–phenotype correlation. They found that patients with LQT1 and LQT2 had a significantly higher risk of a first-time event, higher likelihood for recurrent events, and a younger age of onset, than those with LQT3. Longer QTc interval was found to be an independent risk factor for cardiac events. Although those with LQT1 had the lowest mean QTc interval, those who did have a QTc>500 ms were at the highest risk.Reference Zareba, Moss and Schwartz 14 By 2003, when five genes had been linked to LQTS and the natural history of the disorder was still being explored, Priori et al were among the first to attempt to determine the probability of a first cardiac event in these patients and define the factors that placed them at higher risk. This study of 647 patients examined the incidence of a first-time cardiac event before the age of 40 years in the absence of treatment. Overall, female patients with gene variants at the LQT2 locus and male patients with gene variants at the LQT3 locus had the highest annual incidences. They also found that the QTc intervals of those patients who experienced a cardiac event were significantly longer than those who had not. Interestingly, although LQT1 was by far the most common type in this study, these patients were also at the lowest risk for a first-time cardiac event.Reference Priori, Schwartz and Napolitano 15
The risk of sudden death is not equivalent among all young athletes with LQTS, but rather varies depending upon the genotype and mutation, age, sex, and therapy. Studies have shown that, although females with LQTS in general have a longer QTc interval, males have a higher overall risk for cardiac events.Reference Liu, Jons and Moss 7 , Reference Priori, Schwartz and Napolitano 15 – Reference Hobbs, Peterson and Moss 17 Downregulation of potassium channel genes by female sex hormones have been implicated in this finding of a longer QTc interval among females, but this does not explain the higher risk of clinical events among males.Reference Malloy and Bahinski 18 Studies have demonstrated that, among younger athletes, males with LQT1 are particularly at risk for having a cardiac event; however, after the age of 13, females with LQT2 are at the highest risk. In fact, women with LQT2 continue to have a very high rate of recurrent events following their first episode with 58% having another event within 2 years of follow-up.Reference Liu, Jons and Moss 7
Defining risk for the genotype-positive, phenotype-negative (concealed LQTS) patient has been particularly challenging. A study of 1861 genotype-positive patients found that the QTc interval distribution ranged from 350 to 800 ms with a mean of 450±56 ms, and overall about 25% were within the normal range. This distribution was also similar among the three most common genotypes. Of this large number of genotype-positive patients, 469 had a QTc within the normal range. Consistent with the studies described above, patients with a QTc interval within the normal range had a significantly lower risk (72%) of aborted cardiac arrest or sudden cardiac death, but still had a much higher risk than their genotype-negative family members. Among those with a normal QTc, LQT1 and LQT3 patients were at much higher risk than LQT2 patients, which is in contrast with those with a prolonged QTc interval, as noted above.Reference Goldenberg, Horr and Moss 19
Ultimately, these studies exploring risk stratification have been the cornerstone for consensus guidelines regarding sports participation. As we understand the risks associated with each genotype and mutation based upon the particular derangement in physiology they create, we will be able to more precisely define risk as it pertains to the LQTS patient.
The impact of β-blockade
β-Blockers have been found to greatly reduce the risk of a first-time event in all patients with LQTS, particularly in LQT1 patients (Table 1). As previously described, the main trigger for patients with LQTS, and for athletes in particular, is adrenergic stimulation. At this point, β-blockers are considered standard of care and have been shown to reduce the risk of cardiac events to below 10%.Reference Villain, Denjoy and Lupoglazoff 20 Only one study showed a <90% reduction in risk with full medication compliance and avoidance of QT-prolonging medications in LQT1 patients.Reference Vincent, Schwartz and Denjoy 21 Studies have also suggested that the protective properties of β-blockers continue to be evident in those who have experienced a first-time event, as the risk for subsequent events is still lowered.Reference Liu, Jons and Moss 7 This evidence of high efficacy with β-blockade in preventing cardiac events in LQT1 patients, who are at the highest risk during exercise, is the primary impetus for sports eligibility liberalisation as will be discussed below.
Guidelines for return-to-play
As our understanding of risk stratification, genotype–phenotype correlation, and therapy have improved, so have our guidelines regarding sports participation. The Bethesda guidelines published in 2005 provided a QTc interval “cut-off” for LQTS diagnosis at 470 ms in males and 480 ms in females. These recommendations stated that any patient who met diagnostic criteria, had a history of LQTS-related symptoms, or had an implantable cardioverter defibrillator implanted should be restricted from all athletic participation aside from the Class IA category of sports. These activities included billiards, bowling, cricket, curling, golf, and riflery. These sports are broadly considered to have both the lowest possible dynamic and static components. Although these guidelines were based on the best evidence at the time, the council admitted that their guidelines were more so based on the “art of medicine”.Reference Mitchell, Haskell, Snell and Van Camp 22 , Reference Pelliccia, Zipes and Maron 23 According to these guidelines, however, patients who are genotype positive but do not exhibit even one of these three criteria are considered phenotypically negative and can participate in any athletic activity, except for swimming. The evidence at the time indicated that, although these patients’ risk was not zero, it was low enough to warrant exclusion.
In the same year, the European Society of Cardiology also published consensus recommendations for athletic participation for patients with LQTS. These guidelines were much more rigid than their Bethesda counterparts in that all patients with a diagnosis of LQTS, even those who were phenotypically negative, should be restricted from all forms of athletic participation. Furthermore, the lower end of the QTc interval threshold for moving forward with genetic testing for LQTS was more conservative at 440 ms in males and 460 ms in females. There were no additional recommendations for patients who were genotype negative but with borderline QTc intervals.Reference Pelliccia, Fagard and Bjornstad 24
Our understanding of LQTS has come a long way since these two consensus documents from 2005 were published. It has been demonstrated, as previously mentioned, that certain groups are at higher risk than others, such as males with LQT1 who are younger than 13 years of age and have a QTc>500 and, even more so, females over 13 years of age with LQT2. Beyond such generalisations, however, specific risk to an individual was still very difficult to ascertain. At the same time, it became recognised that by respecting patients’ and families’ autonomy to make an informed decision whether or not to participate in sports, there was a population of patients who could be studied in order to determine the actual risk involved. In 2013, Johnson and Ackerman performed the largest study of athletes with LQTS. They determined that since the 2005 Bethesda guidelines were not firmly grounded in substantial evidence, they would embrace patient autonomy by respecting the family’s decisions to allow their children with LQTS to participate in sports following implementation of a “sudden death safety net strategy”. In total, they had 130 patients with LQTS participating in sports. Of these, none was in compliance with the European Society of Cardiology guidelines, whereas 70 would have been allowed to participate according to the Bethesda guidelines. There were no deaths and only one athlete with LQT1, with QTc interval of >550 ms on follow-up and history of ventricular fibrillation resuscitation, experienced two events. More recently, our group independently examined 212 genotype-positive LQTS patients of whom 103 participated in sports and all of them were treated with β-blockade. There were no tachyarrhythmic deaths, external resuscitations, or syncopal events during sports participation over more than 750 patient years.Reference Aziz, Sweeten and Vogel 25
The HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes in 2013 recommended that all suspected LQTS patients be evaluated by an expert heart rhythm specialist because of the particular difficulty of diagnosing, risk stratifying, and treating this relatively uncommon condition.Reference Priori, Wilde and Horie 26 This recommendation is also partially in response to the increasingly clear evidence since 2005 that universal restriction from sports is likely too conservative, not to mention the numerous consequences of disqualification. With no evidence in the literature that genotype-positive patients without phenotypic manifestations have experienced cardiac events during sports, upholding the European Society of Cardiology-endorsed guidelines for restriction in this population was challenging. Further, a recent study of young athletes demonstrated that they are at particular risk for serious psychological stress due to their diagnoses of potentially lethal cardiac diseases, and removing them from sports likely only exacerbates this underlying vulnerability.Reference Asif, Price, Ewing, Rao, Harmon and Drezner 27
The mounting evidence that patients with LQTS can safely participate as young athletes has resulted in the most recent guidelines put forth in the AHA/ACC scientific statement “Eligibility and Disqualification Recommendations for Competitive Athletes with Cardiovascular Abnormalities: Task Force 10: The Cardiac Channelopathies”.Reference Ackerman, Zipes, Kovacs and Maron 28 First, this document states that all athletes who are suspected to have a cardiac channelopathy and those who have been symptomatic should refrain from sports until being evaluated by a heart rhythm expert. As stated above, as there is no evidence that patients with concealed LQTS are at increased risk for fatal arrhythmias during sports participation, these guidelines state that these patients can universally participate in sports as long as there are precautions in place. These precautions include avoidance of QT-prolonging drugs, electrolyte derangements, and dehydration, while at the same time establishing a safety net of trained providers with access to an on-site automated external defibrillator. The true departure from the 2005 guidelines in this document would allow patients with LQTS who have been symptomatic or have a prolonged QTc interval to return to sports, except swimming for those with LQT1, if they have been asymptomatic for 3 months after initiation of treatment.Reference Ackerman, Zipes, Kovacs and Maron 28 Task Force 10 recognised the tangible and intangible aspects of sports participation and allowed for individualisation of recommendations.
Future of risk stratification and sports participation guidelines
Decades’ worth of studies involving the link between the specific genotypes and the natural history of these patients have evolved our understanding of LQTS. We now know that these gene variants are linked to specific changes at the cellular level that ultimately result in different phenotypic manifestations for each genetic variant. Complicating our interpretation of genotype is the complexity of variable penetrance and modifier genes that are relatively poorly understood. Until we are able to better elucidate this complexity, guidelines will often be too restrictive in some patients and not restrictive enough in others.
The studied efficacy of β-blockade in the treatment of patients, particularly with LQT1, has largely led to the recognition that the psychological and health benefits of sports participation outweigh the potential risk of a cardiac event. In the future, each individual’s risk based on their specific gene mutation, modifying factors, and electrophysiological characteristics of their re-polarisation may be considered together in order to best determine their specific therapy and their ability to participate in sports.
Acknowledgements
None.
Financial Support
This research or review received no specific grant from any funding agency or from commercial or not-for-profit sectors.
Conflicts of Interest
None.
Ethical Standards
The authors assert that all referenced work contributing to this review complies with the ethical standards of biomedical or medicolegal investigation.