



Cardio-facio-cutaneous syndrome is a genetically and clinically heterogeneous syndrome, accompanied by multiple congenital anomalies.Reference Tidyman and Rauen1 Cardiovascular involvement was reported in 75.5% of the patients.Reference Roberts, Allanson and Jadico2
Long QT syndrome is a repolarisation disorder on the electrocardiogram, characterised by a long QT interval, which may cause syncope and sudden cardiac death.Reference Schwartz, Crotti and Insolia3 This report presents a coincidence of congenital long QT syndrome with cardio-facio-cutaneous syndrome in an infant that has not been reported previously.
Case report
A 6-month-old male patient was referred to our department for screening due to his syndromic appearance. The patient had a history of antenatal polyhydramnios and had sparse, slow-growing, curly hair, absent eyebrows with hyperkeratosis, bulbous tip of the nose, low-set ears, and widespread hyperkeratosis throughout the body (Fig 1). The patient had no significant developmental retardation (weight: 6200 g; 10–25 p) and was being followed up for generalised tonic-clonic epilepsy. On cardiac examination, patient had a grade of 1/6, short systolic murmur at the lower left sternal border. He had no findings of heart failure (e.g. tachypnoea, dyspnoea with feeding, failure to thrive, gallop, crackles, hepatomegaly, etc.). In the electrocardiogram of the patient, the rhythm was sinus, heart rate was 130 beats/minute, PR interval was 0.12 second, QRS axis was normal, and there was no T wave abnormality. The QTc distance was calculated as 500–520 ms (Fig 2). His echocardiography revealed 2 mm small ventricular septal defect in perimembranous region, hypertrophy in the interventricular septum (left ventricular free wall 11 mm; z score: +0.02; interventricular septum 8 mm; z score: +1.73), and slight stenosis starting from the valve in the pulmonary artery (p max gradient 25 mmHg, main pulmonary artery 9 mm; z score: −1.24 and right and left branches 5–6mm; z score: −0.61). In his 24 hour Holter electrocardiographic monitorisation, minimum heart rate was 75 beats/minute, maximum heart rate was 150 beats/minute, and mean heart rate was 90 beats/minute during monitorisation. There were rare isolated, monomorphic ventricular extrasystoles that were not exceeding 1% of the whole beats during the day. Also, QTc distance was calculated longer up to 600 ms during 24 hour Holter monitorisation (Fig 3). There was no index case for long QT syndrome in his family, and electrocardiograms of the parents were evaluated as normal. Chromosome analysis was performed to the patient by the genetic department, and the result was 46; XY. Then the patient was analysed with whole exome sequencing from peripheral blood. As a result of DNA sequence analysis, it was determined that there were heterozygous de novo mutations for cardio-facio-cutaneous syndrome (BRAF and GLRA4 mutation) and SCN5a (p.T1250M, c.3749 C>T) for long QT syndrome type 3.
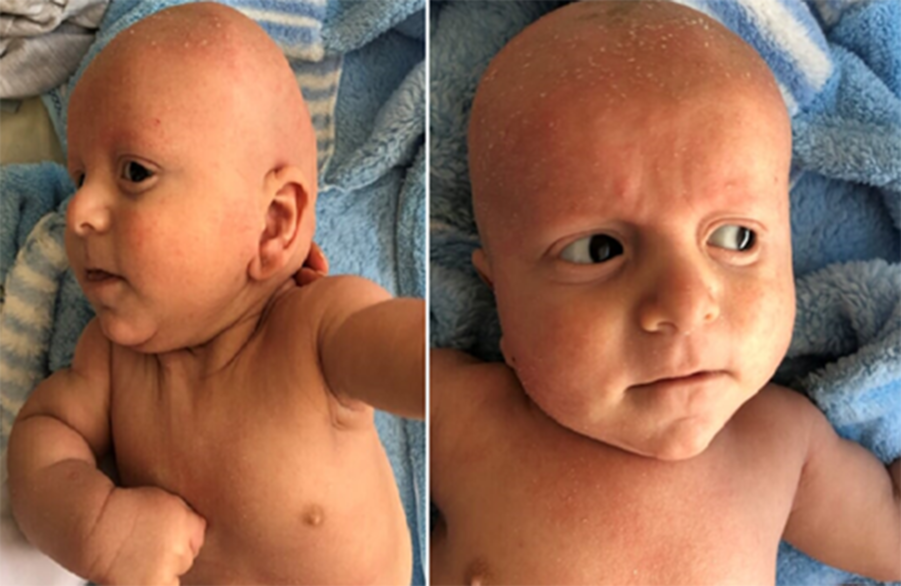
Figure 1. 6-month-old syndromic male patient.

Figure 2. The electrocardiogram sample of the patient.

Figure 3. The 24 hour Holter electrocardiographic monitorization sample of the patient.
The patient, who was asymptomatic cardiologically, started beta-blocker therapy for long QT syndrome. Antiepileptic treatment of the patient was valproic acid, which does not prong the QT interval, was continued, and the patient was included in the regular follow-up programme.
Discussion
The cardio-facio-cutaneous syndrome is one of the RASopathies, caused by gene mutations in the Ras/mitogen-activated protein kinase pathway. Although its frequency is not known all over the world, it has been estimated as 1/810.000 in Japan. Transmission of the syndrome is considered to be autosomal dominant, but occurrence is usually seen sporadic, in most of the cases arising by a new mutation of BRAF, MEK1, MEK2 or KRAS genes, with men and women equally affected.Reference Abe, Aoki and Kuriyama4 The main phenotypic features of the disease are characteristic dysmorphic craniofacial appearance, CHD, dermatologic abnormalities, growth retardation, and intellectual disability and seizures. Cardiac anomalies were reported in 3/4 of the patients. The most common cardiac anomalies are pulmonary valvular stenosis which present in approximately 45% of the patients, followed by atrial septal defect and hypertrophic cardiomyopathy with decreasing frequency. It is known that arrhythmias are not common in cardio-facio-cutaneous syndrome. The arrhythmias reported in the literature were supraventricular tachycardia, atrioventricular block, and Wolff-Parkinson-White syndrome.Reference Allanson, Annerén and Aoki5,Reference Roberts, Allanson and Jadico6 In our case, BRAF and GLRA4 mutation was detected as heterozygous pattern. Patient also had typical craniofacial appearance, dermatologic abnormalities, and epileptic seizure, which are the most common findings of this syndrome. In addition, the patient’s cardiac pathologies, such as pulmonary valve stenosis and septal hypertrophy, were compatible with those common in the disease.
Although arrhythmias are unusual in this syndrome, our patient was first diagnosed with long QT syndrome. The long QT syndrome is a malfunction of cardiac ion channels that cause repolarisation abnormality, recognised by long QT interval in electrocardiography. This cardiac disorder can lead to recurrent attacks of syncope, life-threatening arrhythmias, and sudden cardiac death and may be congenital or acquired. Many genes related to long QT syndrome have been identified so far. KCNQ1 (LQT1), KCNH2 (LQT2), and SCN5A (LQT3) are the most common long QT syndrome genes, accounting for ≈90% of all genotype-positive cases.Reference Kapplinger, Tester and Salisbury7 Long QT syndrome type 3 differs from LQTS1 and LQTS2 in various aspects. Patients present more often with marked resting bradycardia, and QT interval prolongation is more pronounced during slow heart rate. In our case, a heterozygous mutation for SCN5a for long QT type 3 was detected in the genetic examination.
Beta-blockers are used in the management of long QT syndrome, but therapeutic implantation of a cardioverter defibrillator is recommended for patients who have not responded to treatment or had recurrent syncope/cardiac arrest despite treatment as well.
Since our patient had no cardiac symptom, beta-blocker therapy was initiated and the drugs to prolong the QT interval were listed and the family was informed. The patient had no electrocardiographic sign other than prolonged QT interval up to 600 ms, his heart rate was normal for age, had no history of cardiac event, family history of cardiac events, or prolonged QT interval. But the patient is considered as high risk because his QTc interval >600 ms and being long QT type 3.8 Implantation of a cardioverter defibrillator with the use of beta-blockers may be considered for prophylaxis of this patient. We planned to see the patient closely with control 12 lead electrocardiography and to perform repeated 12 channel Holter electrocardiography. In any case of cardiac symptom, we considered to add medications or implantation of a cardioverter defibrillator.
The diagnosis of long QT syndrome has not been previously reported in cardio-facio-cutaneous syndrome in the literature. It should be kept in mind that cardiac examination in cardio-facio-cutaneous syndrome should include electrocardiographic scanning even in early infancy.
Acknowledgements
None.
Financial Support
This research received no specific grant from any funding agency or from commercial or not-for-profit sectors.
Conflicts of Interest
None.
Ethical Standards
The study was carried out in accordance with national and international recommendations, including the Declaration of Helsinki, and has been approved by the local ethics committee. Written informed parental consent was obtained.



