


Just over two decades ago, Roberts wrote a provocative editorial about aortic atresia. He posed the question as to whether, at that time, aortic atresia was the worst heart disease in terms of survival.1 Certainly it was then. Even today, hearts with hypoplasia of the left ventricle represent one of the most serious and challenging forms of congenital heart disease. But a lot has changed for patients with hypoplasia of the left heart since the early 1980s, the experience of Norwood et al. establishing the sequence beginning with staged univentricular palliation, and concluding with creation of the Fontan circulation.2, 3 Despite the once rancorous debate as to whether patients with hypoplasia of the left heart should be subjected to palliative surgery, such discussion has now largely abated, assuaged perhaps by ever improving surgical results. It is justifiable, therefore, to ask which congenital cardiac malformation, or complex of malformations, now deserves the title as “the worst heart disease”? Two combinations spring immediately to our minds. The first is the group of patients with gigantic hearts in the setting of pulmonary atresia with intact ventricular septum and florid tricuspid regurgitation.4 The second is the group of patients with right isomerism.5, 6 In this review, we will focus our attention on the latter complex. As we consider the outcome of patients with right isomerism, also known as the asplenic variant of visceral heterotaxy, it is worth noting that more than four decades ago, Roberts et al.,7 the same Roberts as cited above,1 also wrote: “the finding of asplenia and situs inversus in a patient with congenital heart disease virtually precludes the presence of cardiac lesions which would be benefited by corrective or even palliative surgical procedures”. Let's see if such pessimism is still justified!
Nomenclature
It was just over half a century ago that Bjorn Ivemark,8 while studying at the Children's Hospital in Boston, encountered a number of hearts with complex congenital malformations obtained from patients all of whom also had congenital absence of the spleen, as well as abnormalities of visceral lateralization, with persistence to an unusual degree of symmetry of the thoracic organs.9–12 Following his stellar review, it became customary to describe these patients on the basis of their asplenia, and many still refer to them in this fashion, or simply use the term “Ivemark syndrome” because of his signal contribution. With time, nonetheless, it emerged that some of the patients with these constellations of congenital cardiac malformations could have a normal spleen, a rudimentary spleen, a lobulated spleen, or even multiple small spleens.13–18 Irrespective of the precise splenic arrangement, the patients usually had bronchial symmetry, with bilateral short and eparterial bronchuses,13, 14 a degree of hepatic symmetry,9–12 intestinal non-fixation or malrotation,9–12 bilateral sinus nodes,19–21 and abnormal juxtaposition of the inferior caval vein and the abdominal aorta.22 The arrangement of the abdominal organs in these patients, while sometimes suggestive of the usual or mirror-imaged patterns as assessed using frontal abdominal radiographs, more typically seemed unclear, promoting the suggestion that the overall arrangement was ambiguous.23 Both atrial appendages, nonetheless, were noted to resemble the normal morphologically right atrial appendage, with pectinate muscles extending bilaterally round the entirety of the atrioventricular junctions. Indeed, the morphology of the atrial chambers, particularly the abnormal venoatrial connections, was such to exclude clear-cut and unequivocal identification of either the usual or mirror-imaged arrangements.24–27 Noting the obvious thoracic symmetry, along with a degree of hepatic symmetry, with both lobes of the liver being “right-like” in shape, and coupled with absence of the spleen, some thought these patients exhibited “bilateral right-sidedness”.28 Those who had focused attention specifically on the apparent likeness of the atrial appendages, particularly when using the extent of the pectinate muscles as a marker of morphological rightness or leftness, pointed out that the morphology of the appendages permitted description, in terms of the heart, on the basis of right or left isomerism.24–27 Description of the atrial arrangement on the basis of the morphology of the appendages, of course, did not excuse the need also to determine splenic status. Indeed, an autopsy studied had shown marked variability of the spleen, not only in terms of its presence or absence, but also its variability in size, shape, and function.17 The issue of nomenclature similarly did not obviate the real concern about protecting patients with splenic hypofunction from overwhelming sepsis. There is ample literature about methodologies to define the presence or absence of splenic tissue, the site of such tissue,29 and the need for prophylactic regimes for those with absent or deficient splenic function.30 We should also remember that recognition of the isomeric arrangement has a relatively long history, with “a teratologic syndrome of visceral symmetry” being described over four decades ago,11 and with Van Mierop and Wiglesworth20 demonstrating unequivocal isomerism of the sinus nodes shortly thereafter in patients with absence of the spleen. Despite these early findings, nonetheless, it was more usual to group the patients with the particular combinations of congenital cardiac malformations under the overarching terms of cardiosplenic31 or heterotaxy syndromes.32, 33 “Heterotaxy” itself derives from the Greek roots heteros, meaning other, and taxis, meaning order. Hence, it should strictly account for all other arrangements but the normal, including the mirror-imaged arrangement.34 In this respect, therefore, whilst the patients with right or left isomerism unequivocally exhibit marked degrees of visceroatrial heterotaxy, the term “heterotaxy” does not distinguish those with right from left isomerism, nor the small minority that have a truly mirror-imaged arrangement. And, other than the appearances of the atrial appendages, there is not a single cardiac anomaly, including azygos continuation of the inferior caval vein, that clearly distinguishes right from left isomerism, or asplenia from polysplenia.34, 35 For the purpose of our review, we have analysed the group of patients unified because of isomerism of the morphologically right atrial appendages. The majority, but not all, of these patients will also lack any splenic tissue.36 We are well aware that some authorities disagree with the concept of isomerism, and its use to describe certain forms of atrial arrangement.37, 38 These are the same authorities, nonetheless, who established the importance of the “Morphological method”, which states that one variable feature should not, in the setting of the congenitally malformed heart, be used to define another feature which is itself variable.39 On this basis, venoatrial connections are ruled out for diagnosing atrial arrangement in patients with visceral heterotaxy, since it is the venoatrial connections themselves which are most variable in this setting. The atrial appendages, in contrast, when judged on the basis of the extent of the pectinate muscles, are unequivocally isomeric.27 We should emphasise that it is only the appendages that are truly isomeric. We are not promoting a concept of isomerism of the entire atrial chambers, although it should also be noted that transgenic mice have now been created by knocking out the genes Pitx2 and Cited2, and that these animals show remarkable symmetry of the entirety of the atrial chambers.40, 41 Taken overall, therefore, there can now be little doubt that the concept of right isomerism has a firm scientific foundation.
Types of congenital heart malformations associated with right isomerism
There is a substantial literature devoted to the congenital cardiac malformations associated with right isomerism.5, 6, 8–10, 21, 27, 34, 35, 41–43 These include abnormalities of cardiac position, anomalies of systemic and pulmonary venous connections, complex forms of atrioventricular septal defect, defective ventricular septation, abnormalities of ventriculoarterial connection, and atresia or stenosis of the subpulmonary outflow tract. Hearts have also been described with aortic atresia, or other forms of marked obstruction within the systemic ventricular outflow tract,44–46 but they are uncommon, certainly less common than the frequency of such obstruction in patients with left isomerism.47 Complex forms of pulmonary atresia, in contrast, are frequent in patients with right isomerism, such as the finding of non-confluent pulmonary arteries, each supplied by its own arterial duct.48, 49 The finding of concordant ventriculoarterial connections is distinctly unusual in patients with right isomerism.28 There is, of course, also a wide range of congenital abnormalities not only of the thoracic and abdominal organs, but also the central nervous system.9–14, 50–53 We will discuss these important associations in greater detail below, but it is worth noting that it was the variety in such findings that prompted the designation of the entire spectrum of visceral heterotaxy as “situs ambiguous”.21 If the findings in each system are described separately, and with precision, however, then there is no ambiguity, albeit that all systems do not show harmonious isomeric features.
Extracardiac anomalies and genetics of right isomerism
As emphasized above, those born with right isomerism have a wide range of congenital malformations, involving not only the heart, but also the thoracic and abdominal organs and the central nervous system. This is in marked contrast to the situation found with hypoplasia of the left heart, where relatively few afflicted infants have associated congenital malformations.9–14, 50–53 Ticho et al.51 found that almost half of their patients with absence of the spleen had lesions involving midline structures, most frequently involving the musculo-skeletal or genitourinary systems. Fused adrenal glands, and anal stenosis or atresia, was seen only in patients with absence of the spleen, and not in those with multiple spleens.51 These authors51 published a useful table collating the extracardiac anomalies seen in their large cohort of patients with visceral heterotaxy, divided by them into the subsets of asplenia and polysplenia.
Abnormalities in the right and left patterning of the body have held particular interest for those exploring the fundamental mechanisms of organogenesis, and also for the molecular geneticist.54–58 We have already discussed the major advances made recently in unraveling the sequence of molecular events that produces the lateralization that is characteristic of the normal body plan. A cascade of genes has been implicated, including Pitx2 and Cited2, which have now been knocked out to produce animals with unequivocal right isomerism.40, 41 Other genes, such as Sonic Hedgehog, are believed to produce a midline barrier during normal development, so as to prevent the gene products responsible for producing morphological leftness to reach the right side of the developing body. Several of these, and other, genes have now been implicated in the development of heterotaxy, and its associated congenital cardiac malformations, in the human.54–61 The findings suggest that, while it is possible that heterotaxy can result because of mutation in single genes, extensive heterogeneity of locus is likely.54 Genes implicated in human heterotaxy include Zic3, LeftyA, Cryptic, and Acvr2B. Nkx2.5 and Crelda may also have a role in abnormal lateralization. Because heterotaxy is also known to be caused by exposure to teratogens, such as retinoids,62–64 at least in the experimental animal, it is also suggested that the complex pattern of inheritance likely results from a combination of teratogenic effects and single gene disorders, with variable expression and incomplete penetrance.54 Chromosomal abnormalities, while reported on occasion in patients with right isomerism, are uncommon, and do not seem specific for heterotaxy. From clinical experience, it seems that while most examples of right isomerism occur as sporadic events, there are numerous examples of familial occurrence in siblings. Depending on the particular family, these findings suggest either an autosomal dominant, a recessive, or an X-linked pattern of inheritance.35, 65–73 There is one remarkable family in which four siblings lacked any splenic tissue.74 Some years ago, it was suggested that there appeared to be an increased incidence of the complex cardiac malformations associated with isomerism amongst the Muslim Asian population.75 We are not aware of any further studies confirming or refuting this observation.
Prevalence of right isomerism
It has been estimated that heterotaxy, or at least the cardiac malformations associated with right or left isomerism, occurs in from 1 to 1.44 in each 10,000 livebirths.70, 76, 77 In most studies, a ratio of around 2 males is noted for each afflicted female.5, 6, 76, 77
Outcomes for patients with right isomerism
Fetal diagnosis of isomerism of the right atrial appendages is now firmly established and well-documented.6, 31, 78–86 Thus, Berg et al.31 argued that right isomerism should always be suspected in the fetus exhibiting a combination of at least two of the findings of atrioventricular septal defect with common atrioventricular valve, juxtaposition of the inferior caval vein and the descending aorta, and abnormal arrangement of the thoraco-abdominal organs. It would seem, in fact, that this is somewhat conservative, since the juxtaposition of the caval vein and aorta is generally taken to be diagnostic for right isomerism in postnatal life. Sharland and Cook82 found that, in fetal life, left isomerism is diagnosed twice as often as right isomerism, a finding endorsed by the recent study of Lim et al.6 Spontaneous fetal loss occurs far more frequently with left than with right isomerism, likely attributed to the heart block and hydrops known to accompany many cases of left isomerism diagnosed during fetal life. The incidence of termination of pregnancy varies greatly, according to sociocultural, religious and ethical issues, as well as inherent biases for institutional and individual counseling. In the experience reported by Sharland and Cook,82 slightly more than half of families chose to terminate their pregnancy once the diagnosis was made for isomerism. Surveys of the literature also reveal termination of a significant number of pregnancies following the diagnosis of complex forms of atrioventricular septal defect,87 right-sided heart,88 or totally anomalous pulmonary venous connections,82, 89–91 all conditions known to be associated with isomerism. In a contemporary review of 178 cases diagnosed with heterotaxy syndromes at the Toronto Hospital for Sick Children between 1990 and 2003,6 right isomerism was found in 75 cases (42%), with a prenatal diagnosis having been made in 33 of these. While the spectrum of cardiac and extracardiac anomalies did not significantly differ among the patients diagnosed before or after birth, the pregnancy was terminated in two-fifths of those diagnosed antenatally. In a further 17%, the parents elected for non-intervention after delivery, with all of these patients dying. This review also demonstrated better postnatal survival amongst those with left isomerism compared with those with right isomerism,6 endorsing the previous observations published by Hashmi et al.5 and Gilljam et al.,47 again using the Toronto database.
When considering the potential role of cardiac surgery for patients diagnosed with right isomerism, it is possible again to draw parallels or comparisons with the evolution of surgery for hypoplasia of the left heart. Appreciating that the outcome for those born with hypoplastic left heart syndrome was, with rare exceptions, uniformly fatal in early infancy coincident with ductal closure,92 two factors contributed importantly to the impetus to salvage babies with gross underdevelopment of the left heart. The first was the introduction of the Fontan operation in 1971,93 an operation designed both to bypass the right heart and to separate the systemic from the pulmonary venous circulations in patients with tricuspid atresia. The second was the establishment of the New England Regional Infant Cardiac Program.94 This latter study, published in 1980, characterized babies with hypoplasia of the left heart as being typically born at full-term, and for the most part being free of other important cardiac and non-cardiac congenital abnormalities. Furthermore, most had normal chromosomes. The matter of chromosomal make-up has been the subject of some more recent discussion and debate.95, 96 Once the Fontan procedure was applied to forms of congenital heart malformations other than tricuspid atresia with a dominant left ventricle, or some other form of univentricular atrioventricular connection, an obvious extension of the procedure was for the patient with hypoplasia of the left heart.97 The remarkable surgical results now achieved in the treatment of patients with the hypoplastic left heart syndrome, whether following the path of univentricular palliation or cardiac transplantation, have been documented elsewhere.97–104 What can be said, then, of patients with the complex congenital cardiac malformations seen in the setting of right isomerism?
Considering the complexity of these malformations, it is not surprising that the outcome for many is so poor. A few individuals with severe cardiac disease and right isomerism are catalogued as experiencing moderate longevity with or without palliation, but such occurrences are rare.105–108 For the overwhelming majority, survival requires surgical intervention. Just as some patients with aortic atresia are candidates for a biventricular surgical repair, the same can be said for some patients with right isomerism. When analyzing the larger series of patients reported with right isomerism,109–114 the majority are potential candidates only for univentricular palliation or cardiac transplantation.5, 6, 8–10, 13–15, 21, 27, 34, 35, 37, 41 Having said that, there is also no doubt that the outcomes in recent years have dramatically improved for patients with both right or left isomerism who require functionally univentricular palliation, with some centres now reporting surgical mortalities of less than 5%, a considerable improvement over earlier studies.115–120 Stamm et al.,117 reviewing the experience of the Boston Children's Hospital, stated that the early mortality after the Fontan procedure in patients with heterotaxy was 19% before 1993, 3% since 1991, and that no patient had died over the period from 1993 to 2000. The results from Toronto, published in 2001,115 while also showing improvement, were not quite as exemplary as the experience in Boston.117 Because many patients with right isomerism have a common atrioventricular valve, often with a morphologically right ventricle functioning as the systemic ventricle,5, 6, 8–10, 13–15, 21, 27, 34, 35, 37, 41, 121 it is not surprising that, in some, progressive atrioventricular valvar regurgitation becomes a threat to a successful outcome following the Fontan procedure. This observation, however, is not universally shared.122 There is increasing experience with repair or replacement of the common atrioventricular valve, or other manoeuvres performed either at as staging procedures or at the Fontan operation, with improving results.111, 123–126 The many anomalies of systemic venous connection seen with right isomerism can usually be dealt with at the time of staging, or by modifications of the Fontan procedure, including intra-atrial baffle or diversion procedures.115–120, 127–130 Yet most of the studies reported thus far address only those patients surviving to staging, or those undergoing the Fontan operation or one of its many modifications. What is not generally discussed is the attrition of these patients from birth through the rigours of early univentricular palliation. Several institutions have attempted to provide more comprehensive data about outcomes. Franklin et al.,131–133 in their exhaustive examination of a large cohort of 191 infants with double inlet ventricle, provided striking evidence of the poor prognosis of infants with right isomerism. Of their patients who had right isomerism, the survival at 2 years was no more than 17 plus or minus 6%, with survival at 5 years falling to 14 plus or minus 6% (Fig. 1).
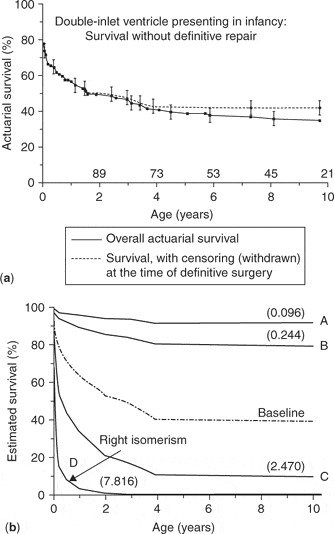
Figure 1. (a) Overall survival of infants with double inlet ventricle without surgical intervention. (b) Predicted survival according to the morphology of the dominant ventricle. A: usual atrial arrangement, double inlet left ventricle, discordant ventriculoarterial connections, valvular or subvalvar pulmonary stenosis with balanced pulmonary blood flow; B: the same as “A” but with low pulmonary blood flow; C: usual atrial arrangement, double inlet left ventricle and single outlet aorta; D: patients with right isomerism, double inlet and double outlet right ventricle, common atrioventricular valvar orifice, single outlet with pulmonary atresia, totally anomalous pulmonary venous connections and low pulmonary blood flow, presenting less than 14 days of age. Reproduced from Ref. 131, Franklin et al., with permission of the Journal of Thoracic and Cardiovascular Surgery.
There are other studies also supporting the overall dismal survival of young infants with right isomerism.5, 6, 134 Rose et al. from Toronto showed 30 years ago that the survival at one year for patients with right isomerism was only 21%.42 The outcomes for patients with right isomerism is considerably poorer than for those with left isomerism, despite the fact that the two groups share many similar cardiac anomalies.5, 6, 38, 47
For those requiring univentricular palliation, several anatomic features seemingly influence surgical outcome and attrition. For example, Cheung et al.135 found that more severe obstruction to the subpulmonary outflow tract worsens the surgical outcome (Fig. 2).
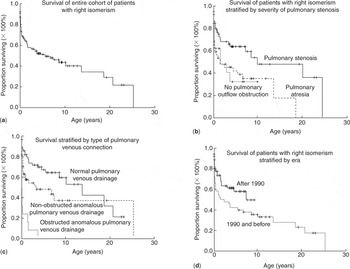
Figure 2. (a) Kaplan-Meier estimate of survival for the entire cohort. (b) Kaplan-Meier estimates of survival by different types of pulmonary venous drainage. (c) Kaplan-Meier estimates of survival by different types of pulmonary outflow tract obstruction. (d) Kaplan-Meier estimates of survival by different era. Reproduced from Ref. 135, Cheung et al., with permission of Heart.
Their data also showed that those patients without any obstruction in the pulmonary outflow tract, admittedly a rare finding in those with right isomerism, also fared poorly, likely reflecting coexisting severe obstruction to the systemic ventricular outflow tract. Data reviewing the natural and unnatural history of patients with pulmonary atresia136 also showed that those with right isomerism did very poorly, with less than 13% survival. Those patients gaining the most surgical attention and concern have been those with both severe obstruction of the pulmonary outflow tract obstruction and anomalies of pulmonary venous connection, in addition to the other associated complex cardiac anomalies of right isomerism. Such young neonates, requiring not only creation of a systemic-to-pulmonary shunt to augment pulmonary blood flow, but also repair of obstructed totally anomalous pulmonary venous connections, have had, with few exceptions, a very grim outlook. Phoon and Neill,137 in their retrospective review of patients described by others, noted that totally anomalous pulmonary venous return and pulmonary atresia were both significant risk factors for early mortality. For the 191 patients in whom they were able to gather pertinent data, median survival was no more than 2.6 months137(Fig. 3).

Figure 3. Predicted survival curve of patients with right isomerism based on age at death from reported cases in literature. Reproduced from Ref. 137, Phoon and Neill, with permission of the American Journal of Cardiology.
Their study, despite being biased by its methodology, reflects the poor prognosis for patients with right isomerism, since similar conclusions were reached from a number of studies made using the experience of single institutions, such as those of Sadiq et al. from Birmingham, England,134 Hashmi et al. and Lim et al. from Toronto,5, 6 and Gaynor et al. from Philadelphia.138 Totally anomalous pulmonary venous connection, of course, is a necessity in the setting of right isomerism,5, 6, 8–10, 13–15, 21, 27, 34, 35, 37, 41 since both atrial chambers possess a morphologically right appendage. Thus, even if the pulmonary veins return to the atrial chambers, they must do so in morphologically abnormal fashion. The site of the anomalous pulmonary venous return is varied, and includes unilateral or bilateral anomalous drainage, which may or may not be obstructed. When there is important obstruction to pulmonary venous flow, the severe reduction in the rate of flow may mask the clinical importance of the pulmonary venous anomaly.139–141 So as to combat this potential problem, we devised the prostaglandin challenge141 in the era before the routine application of cross-sectional echocardiographic imaging. The test was designed to augment pulmonary blood flow in order to unmask any concealed pulmonary venous obstruction. More recent data now suggests that such masked pulmonary venous obstruction may be less common than once thought.142 Failure to define the arrangement of the pulmonary veins preoperatively, nonetheless, results in high surgical mortality, with the findings reported in 2000 by Chowdhury et al.143 providing validation for the concern expressed much earlier by DeLeon et al.144 These observations are further supported by the experience reported by Jenkins et al.,145 showing that, in the setting of totally anomalous pulmonary venous connection, a small sum of the diameters of the individual pulmonary veins, a small confluence, and the presence of heterotaxy syndrome were all significant predictors of survival in univariate analysis. Multivariate analysis showed that the small size of the pulmonary veins was a strong adverse predictor of survival independent of the presence of heterotaxy. The patients with heterotaxy syndrome, however, had significantly smaller pulmonary veins than those with usual atrial arrangement.145 Their findings were subsequently confirmed by Kawahira et al.,146 who calculated a pulmonary venous index. Combining this index in a formula incorporating the mean difference in pressure between the pulmonary arteries and the left atrium and pulmonary veins showed that the measurements were indicative of outcome in a small series of patients undergoing cavopulmonary shunting or the Fontan procedure.146
The experience reported by Sadiq et al.134 also attests to the deleterious effects of the need for both a systemic-to-pulmonary arterial shunt and relief of pulmonary venous obstruction. Albeit in a small group of patients, results were much worse for those needing intervention in the first month of life because of obstructed pulmonary venous return combined with pulmonary atresia. All 5 of their patients in this group requiring a shunt and surgical relief of pulmonary venous obstruction died (Fig. 4). In contrast, survival was very much better in those not requiring surgery until later in life, with only a small proportion of this group having the insidious combination of obstructed pulmonary venous return and pulmonary atresia.

Figure 4. The two curves show depicted survival of patients with right isomerism depending on age at initial operation. Reproduced from Ref. 134, Sadiq et al., with permission of Heart.
More evidence is provided by the appreciably larger experience reported by Gaynor et al.138 for the period between 1984 and 1997 (Fig. 5). They analysed the results for all patients having totally anomalous pulmonary venous connection in the setting of the functionally univentricular heart, with over two-thirds of the cohort also having isomerism. The overall survival for their cohort was 45% at 6 months of age, 37% at 1 year of age, and 19% at 5 years. Of their patients, 12 died without any surgery. Of the 61 patients undergoing surgery, survival was 54% at 6 months of age, 44% at one year of age, and 23% at 5 years. For their entire cohort, survival was worse for those with obstructed pulmonary venous connections and, as reported by Sadiq et al.,134 repair of the anomalous pulmonary venous connections at the time of the initial operation was associated with worse survival.
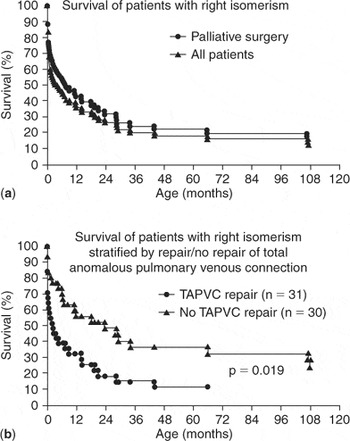
Figure 5. (a) Actuarial survival curve for the entire cohort compared to those who underwent staged reconstruction. (b) Actuarial survival curves comparing those who underwent repair of total anomalous pulmonary venous connection with those who did not have pulmonary vein surgery. Reproduced from Ref. 138, Gaynor et al., with permission of the Journal of Thoracic and Cardiovascular Surgery.
The review of Cheung et al.,135 which we have already discussed in the setting of obstruction to the subpulmonary outflow tract, also addressed the influence of obstructed pulmonary venous connections. They had reviewed 116 patients seen between January 1980 and December 2000 at the Grantham Hospital in Hong Kong (Fig. 2). No interventions were planned in 31 patients (27%), and all of these died. For the remainder, mean survival at one month, 1, 5, 10, and 15 years was 80%, 65%, 51%, 43%, and 34%, respectively. The significant adverse independent risk factors for survival were pulmonary venous obstruction, and functionally univentricular morphology. Further evidence of the malignant influence of obstructed pulmonary veins is provided by the analysis made in 19985 of the children with right isomerism included in the database of the Toronto Hospital for Sick Children; including some of the patients reported earlier by Rose et al.42 The review made in 1998 concentrated on the management and outcomes for the 91 patients with right isomerism seen over the period from 1970 to 1996 (Fig. 6). Of this cohort, nine-tenths had presented within the first month of life, with almost two-thirds being already recognized at birth. As anticipated, the cardiac malformations included an atrioventricular septal defect with common valve in four-fifths, functionally univentricular arrangement in almost three-quarters, abnormal ventriculoarterial connections in all but one-twentieth, atresia or stenosis of the subpulmonary outflow tract in five-sixths, and overtly anomalous pulmonary venous connections in almost nine-tenths. Even for those considered to have “normal” pulmonary venous connections at this time, however, we now appreciate that, because of the presence bilaterally of morphologically right atrial appendages, the pulmonary venous connections must have been anatomically abnormal in all. Irrespective of this nicety, the pulmonary venous connections were noted to be obstructed in three-tenths of the cohort. Overall mortality for the group was 69%. No interventions had been either planned or performed in one-quarter of the cohort, and 95% of these died. For those requiring their first cardiovascular operation in the neonatal period, mortality was 75%, as opposed to 51% for those not needing operations until later, with this difference being statistically significant. Of those needing both repair of the pulmonary veins and construction of a systemic-to-pulmonary arterial shunt in the first month of life, nineteen-twentieths died.
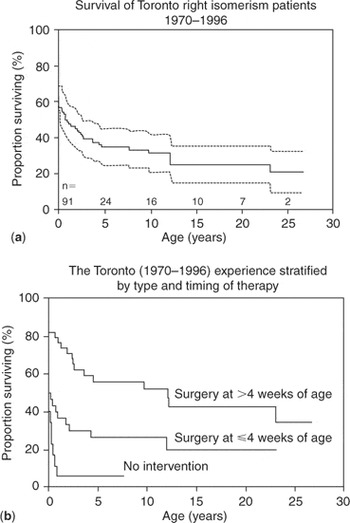
Figure 6. (a) Actuarial survival curve for entire cohort of patients with right isomerism seen from 1970 to 1996 at the Toronto Hospital for Sick Children. (b) Kaplan-Meier survival curves dpending on type and timing of therapy. With no therapy, 95% mortality; with initial surgical intervention as a neonate, 75% mortality; and with initial surgical intervention after 4 weeks of age, 51% mortality. Reproduced from Ref. 5, Hashmi et al., with permission of the Journal of the American College of Cardiology.
Overall estimates of survival were 71% at 1 month, 49% at 1 year, and 35% at 5 years. Independent risk factors for decreased time to death included an unobstructed pulmonary outflow tract, severe atrioventricular valvar regurgitation, and obstructed pulmonary veins. Although the majority of the patients were candidates for functionally univentricular palliation, less than one-quarter achieved completion of the Fontan circulation, with a surgical mortality of 21%. This experience was then endorsed by another analysis of the Toronto material. Thus, Calderone et al.147 showed that repair of totally anomalous pulmonary venous connections in the setting of the functionally univentricular heart was associated with less than a 30% long-term survival, whereas mortality for repair of patients with uncomplicated totally anomalous pulmonary venous connections decreased from 26% to 8% over the same period. They also highlighted the malignant influence of the need for both banding of the pulmonary trunk and repair of pulmonary veins in those with the functionally univentricular circulations.147 As a comparison, we can also look at the outcomes of the patients with left isomerism in the same analysis of the Toronto experience (Fig. 7). Survival is obviously better for those with left isomerism, albeit that the ones requiring univentricular palliation still fared poorly (Fig. 7a).
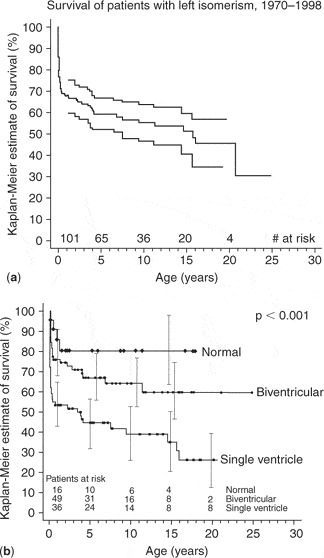
Figure 7. (a) Actuarial survival of patients with left isomerism. (b) This graph depicts predicted survival for those with a normal heart, those undergoing a biventricular or single ventricle repair. Reproduced from Ref. 47, Gilljam et al., with permission of the Journal of the American College of Cardiology.
We have discussed above some relatively recent experiences reported from the Children's Hospital of Boston. More than a decade ago, however, Heinemann et al.148 reviewed the outcomes of for their patients with heterotaxy and totally anomalous pulmonary venous connection, and produced a somewhat more rosy picture. We should again caution that the Boston group do not consider pulmonary veins returning to the heart in the setting of “asplenia” to be anatomically abnormal, irrespective of whether or not the patients have appendages bilaterally of right morphology. Be that as it may, over the period from 1981 and 1991, they encountered 38 patients with heterotaxy needing palliation within the first 3 days of life, and in 21 of these, they identified totally anomalous pulmonary venous connections, with 18 having extracardiac connections to a systemic vein. In two-thirds of this group, the connections were obstructed, and the patients needed an emergency operation. When univariate analysis was performed for the overall group, neither the presence of totally anomalous pulmonary venous connection, nor its repair in isolation or with shunting, emerged as a definite risk factor for early death. When a shunt was performed as part of the first operation, however, survival was improved. They observed, nonetheless, that the need for a concomitant shunt in patients with obstructed totally anomalous pulmonary venous connection could initially be underestimated. As far as we are aware, the group from Boston has not reported more recent experience with population of patients. Other occasional successes have been reported in repair of the pulmonary venous anomaly without the use of cardiopulmonary bypass.149, 150 Others, drawing an analogy to the management of hypoplasia of the left heart, have measured mixed venous oxygen saturation as a means of estimating the ratio of pulmonary to systemic flows.151 There is also some gratifying experience with cardiac transplantation,152–155 but the numbers of patients benefiting from this approach are very small. And, even for those who survive the initial neonatal palliation, there is ongoing attrition related to occlusion of the shunt, severe cardiac arrhythmias, and overwhelming sepsis156–159 (Fig. 8).
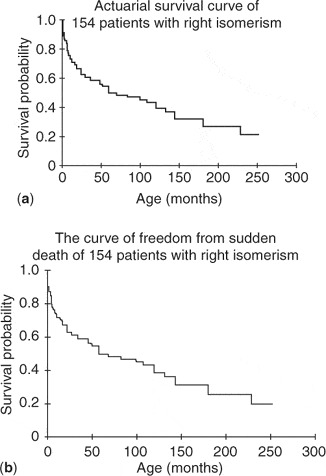
Figure 8. (a) Actuarial survival curve for 154 patients with right isomerism. (b) The curve showing freedom from sudden death of the same cohort as depicted in the upper panel. Reproduced from Ref. 159, Wu et al., with permission of the Journal of Pediatrics.
Conclusions
For our conclusion, we can do no better than to return to the most recent review of the extensive experience at the Hospital for Sick Children, Toronto.6 For the period between 1990 and 2004 (Fig. 9), the estimates for survival at 1 month, 1 year, and 5 years for those diagnosed prenatally and liveborn with right isomerism were 47%, 20%, and 20%, respectively. This can be compared to figures of 88%, 44%, and 31% for those born and diagnosed postnatally, showing that antenatal diagnosis did not improve overall survival, despite facilitating perinatal care. More than three-fifths of patients with isomerism referred to the Toronto centre are currently diagnosed prenatally, and a further rise in this rate of prenatal detection is likely in coming years. When parents opted for an active postnatal management, the figures for survival at 1 month, 1 year, and 5 years improved somewhat, to 74%, 50%, and 40% respectively, but the statistics continue to illustrate the sombre perspectives associated with a diagnosis of right isomerism, despite the progress made in surgery on the pulmonary veins and functionally univentricular palliation. Moreover, none of the survivors underwent a successful biventricular repair, and sadly, we were unable to demonstrate any improvement in outcome during the 14 years of the study. We are impressed with the striking similarity of the estimates for survival of patients with right isomerism published from different world-wide institutions. With only occasional exceptions, these patients continue to have a very poor prognosis. Those requiring surgical intervention in the first month of life certainly qualify as a group with one of the worst forms of congenital cardiac disease.

Figure 9. Actuarial survival of patients with right or left isomerism, comparing outcomes in those diagnosed prenatally to those with postnatal outcome. The upper line for both left and right isomerism gives the data for patients diagnosed postnatally. Reproduced from Ref. 6, Lim et al., with permission of the American Heart Association.
Acknowledgement
Professor Anderson is supported by grants from the British Heart Foundation together with the Joseph Levy Foundation. Research at the Institute of Child Health and Great Ormond Street Hospital for Children NHS Trust benefits from R&D funding received from the NHS Executive.









