


Alveolar capillary dysplasia is a congenital pulmonary vascular abnormality characterised histologically by a lack of formation and ingrowth of alveolar capillaries leading to a failing air–blood barrier first described by Janney et al in 1981.Reference Janney, Askin and Kuhn 1 Alveolar capillary dysplasia is mostly associated with misalignment of the pulmonary vessels as described by Wagenvoort in 1986.Reference Wagenvoort 2 Since then, more than 200 cases of infants, including some cases within families, have been described. This pulmonary vascular developmental disorder is associated with congenital non-lethal malformations in almost 75% of reported cases such as gastrointestinal, urogenital, and ocular abnormalities. In about 10%, alveolar capillary dysplasia is associated with congenital heart disease.Reference Janney, Askin and Kuhn 1 , Reference Rutledge and Jensen 3 – Reference Zufferey, Martinet and Osterheld 19
The clinical picture is dominated by progressive cyanosis in an infant with signs of respiratory distress because of severe neonatal pulmonary arterial hypertension. The outcome is always fatal even if prolonged survival for several months has been reported.Reference Shehata and Abramowsky 11 , Reference Ahmed, Ackerman, Faught and Langston 20 All aggressive intensive care treatment strategies such as mechanical ventilation with 100% oxygen and nitric oxide, pulmonary vasodilators, and even extracorporeal membrane oxygenation are unsuccessful. The definite diagnosis of alveolar capillary dysplasia can only be established by lung biopsy or autopsy. Infants can be admitted directly to a neonatal intensive care unit or to a Pediatric Cardiology Department because of the initial clinical picture of cyanosis with or without predominant signs of respiratory distress, especially if congenital heart disease is suspected or associated. In the case of congenital heart disease, diagnosis can be even more challenging to the clinician as the patient might present a rather atypical clinical course. Until 2008, genetic data on this fatal disease were scarce. Recent reports have identified microdeletions of the forkhead box transcription factor gene cluster on 16q24, which have been linked to some cases of alveolar capillary dysplasia, with and without congenital heart disease, and including other congenital malformations.Reference Stankiewicz, Sen and Bhatt 17
In this report, we review the data of two new patients with alveolar capillary dysplasia and associated congenital heart disease, as well as the literature in the light of these recent genetic data.
Methods
We retrospectively reviewed our institutional records of all clinical and laboratory data available from 2005 to 2010 on all infants admitted to our Pediatric Cardiology Department for whom the final histological diagnosis of alveolar capillary dysplasia was established. We excluded four patients with alveolar capillary dysplasia without cardiac defect, who were only seen for cardiac evaluation before being admitted to our institutional paediatric intensive care department or before being transferred to the regional centre for extracorporeal membrane oxygenation support. We searched PubMed for all articles in English concerning alveolar capillary dysplasia and congenital heart disease. Search terms consisted of “alveolar capillary dysplasia”, “congenital alveolar capillary dysplasia”, “misalignment of pulmonary vessels”, “congenital alveolar dysplasia” associated with “congenital heart disease”, “cardiac malformation”, “heart disease”, “cardiac defect”, “atrioventricular septal defect”, “left heart obstruction”, and “obstructive left heart disease”.
Results
We identified two patients with alveolar capillary dysplasia and associated congenital heart disease. Of these patients, one had a prenatal diagnosis of cardiac malformation in the second trimester of pregnancy.
The first infant was born at 41 weeks of gestation and had a birth weight of 3460 g with an APGAR score of 5–8–9. She presented refractory cyanosis without initial respiratory distress at 2 h of age and was transferred to our Pediatric Cardiology Department because of suspected congenital heart defect. Echocardiography showed an atrioventricular septal defect with a large atrial and ventricular component with a right-to-left shunt via ventricular septal defect and patent arterial duct. This suprasystemic pulmonary arterial hypertension was discordant with the usual haemodynamics of this defect in a newborn. Despite aggressive management of pulmonary hypertension, the child deteriorated and was started on extracorporeal membrane oxygenation. With the parents’ informed consent, the infant was withdrawn from circulatory support once a definite diagnosis of alveolar capillary dysplasia was established by lung biopsy (Fig 1) and she died. Chromosome analysis, initially performed because of the heart defect, revealed a balanced reciprocal translocation between chromosomes 1 and 16. The karyotype was designated as 46,XX,t(1;16)(q32;q24)dn. The balanced rearrangement arose de novo as parental karyotypes were normal. An Agilent 244K oligonucleotide microarray (Agilent Technologies, Santa Clara, California, United States of America) with a 10 kb resolution was performed and no chromosomal imbalances were detected. The 16q24 breakpoint of the patient was located ∼60 kb upstream of the FOXF1 gene (Fig 2), as seen by fluorescence in situ hybridisation analysis using bacterial artificial chromosome clone CTD-2327J16 that showed split signals on both der (1) and der (16) chromosomes with almost equal signal intensity (Fig 3). Supplementary Table S1 shows an overview on all employed bacterial artificial chromosome clones.
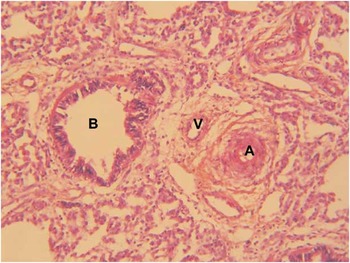
Figure 1 Histopathology of lung biopsy of the first infant showing alveolar capillary dysplasia with misalignment of the pulmonary veins. Haematoxylin and eosin staining, 20×. The pulmonary arterioles (A) are adjacent to pulmonary thin-walled veins (V) and bronchioles (B). The arterioles have increased muscular wall thickness with several layers of muscular cells. The lobular pulmonary architecture is altered with simplified enlarged alveoli and thickened lobular septa.

Figure 2 Schematic representation of the genomic region involving the FOXF1, FOXC2 and FOXL1 genes. The breakpoint (represented by the dotted line) of the balanced translocation t(1;16)(q32;q24) on chromosome 16 lies within the bacterial artificial chromosome CTD-2327J16 approximately 60 kb upstream of FOXF1 gene.

Figure 3 Fluorescence in situ hybridization (FISH) of the affected infant harbouring the translocation t(1;16)(q32;q24). FISH analysis shows split signal using CTD-2327J16 clone (labelled in green) on both der(1) and der(16) chromosomes (red and blue arrows respectively). The white arrow indicated the normal chromosome 16.
The second patient had a prenatally diagnosed ventricular asymmetry with the potential of aortic coarctation and a muscular ventricular septal defect, as well as a left renal duplicity associated with a mega-ureter. Prenatal karyotype was normal and fluorescence in situ hybridisation analysis for 22q11 was negative. His mother had been operated in the neonatal period because of an aortic coarctation. He was born at 40 weeks of gestation with a birth weight of 3960 g and an APGAR score of 1–3–9. The infant became tachypnoeic starting from the second week of life. Postnatal echocardiography of the infant confirmed suprasystemic pulmonary arterial resistances with ventricular septal defect shunting right-to-left in systole but no aortic coarctation was seen. This unusual haemodynamic finding, without other perinatal risk factors of neonatal pulmonary arterial hypertension, motivated cardiac catheterisation. Cardiac catheter demonstrated isosystemic pulmonary arterial hypertension non-reactive to oxygen and nitric oxide without any other cardiac structural anomalies other than ventricular septal defect and patent arterial duct. The suspected diagnosis of alveolar capillary dysplasia was confirmed by lung biopsy. After parental consent, the decision of compassionate management was taken and no further aggressive therapy was initiated. The patient was discharged from hospital and died at home at 6 weeks of age. DNA samples for FOXF1 gene defect analysis were not available in this case.
Literature review
Including Janney's first report, 19 papers concerning 29 infants with histologically confirmed alveolar capillary dysplasia, mostly with misalignment of the pulmonary veins and cardiac defects, were published in English since 1981. Table 1 describes the detailed clinical characteristics of all so far reported patients, including the two infants presented here, concerning congenital heart disease, associated malformations, available genetic data, and the way histological diagnosis was obtained.
Table 1 Characteristics of all published patients including this series, their respective congenital heart disease and mode of diagnosis.

ACD/MPV = alveolar capillary dysplasia/misalignment of pulmonary vessels; ASD = atrial septal defect; AVSD = atrioventricular septal defect; CoA = coarctation of the aorta; CS = coronary sinus; DORV = double outlet right ventricle; HLHS = hypoplastic left heart syndrome; LSCV = left superior caval vein; LV = left ventricle; PDA = persistent arterial duct; PS = pulmonary stenosis; PVR = pulmonary venous return; VSD = ventricular septal defect
The only suggested four cases of ACD/MPV without confirmation at autopsy or biopsy are written in italics
The two predominant heart conditions associated with alveolar capillary dysplasia and accounting for 64% of patients reported in the literature are either various degrees of left heart obstruction or the atrioventricular septal defect. Severity of left heart obstruction ranges from coarctation of the aorta up to hypoplastic left heart syndrome with mitral and aortic atresia. Variants of obstructive left heart disease were encountered in 11 (35%) of the 31 reported patients. Atrioventricular septal defect in its partial or complete form was reported in 9 (29%) of 31 patients. The published cases are variably characterised by a small left ventricle with or without consequent aortic coarctation. Minor heart defects such as persistent arterial duct, right aortic arch, retroesophageal subclavian artery, left superior caval vein to the coronary sinus, atrial septal defect were reported in 6 (19%) of 31 patients. Only in two cases (6%) there was obstruction of the right heart (tetralogy of Fallot, pulmonary stenosis).
In eight (26%) of the 31 published patients, FOX transcription factor gene cluster defects, mostly microdeletions in chromosome 16q24.1q24.2, have been identified. Of these eight patients, three had variants of atrioventricular septal defect, two had patent arterial duct, one infant had hypoplastic left heart syndrome, one had double-outlet right ventricle with pulmonary atresia and small left ventricle, and one had tetralogy of Fallot. The associated congenital malformations, if encountered, appear in Table 1. Genetic data on this recently described microdeletion are not available in the patients reported before 2009 as it has been described only then. There were four patients initially reported by Sen et alReference Sen, Thakur, Stockton, Langston and Bejjani 10 who were retrospectively genetically analysed. In one of them, a FOXF1 mutation was identified and published by Stankiewicz et al.Reference Stankiewicz, Sen and Bhatt 17
In four additional patients with cardiac defects reported in the literature, the diagnosis of alveolar capillary dysplasia has only been suggested by the authors because of the clinical course of the pulmonary disease completely refractory to any treatment modality but permission for lung biopsy or autopsy was declined.Reference Lane, Siwik, Preminger, Stork and Spector 7 , Reference Stankiewicz, Sen and Bhatt 17 One of these children was a foetus who was terminated at 22 weeks of gestation after prenatal diagnosis of hypoplastic left heart syndrome and a heterozygous deletion of chromosome 16 was detected. There were two infants who had atrioventricular septal defect and one infant who had an interrupted aortic arch. Data concerning these patients have been included in Table 1 in italics.
Discussion
The first but only minor cardiac abnormality (retro-oesophageal right subclavian artery) in an infant who died of alveolar capillary dysplasia was reported by Janney et al.Reference Janney, Askin and Kuhn 1 Langston described the typical clinical fatal course of an infant who had a divided but apparently non-obstructive left atrium at autopsy.Reference Langston 4 Despite these earlier reports, the difficulty of the joint diagnosis of alveolar capillary dysplasia and congenital heart disease was first recognised in 1998.Reference Garola and Thibeault 6 Garola and ThibeaultReference Garola and Thibeault 6 reported the cases of two infants with congenital heart disease – coarctation of the aorta and double-outlet right ventricle with hypoplastic left ventricle – who had a rather unusual clinical course in the neonatal period and were diagnosed with alveolar capillary dysplasia at autopsy. Owing to the fact that alveolar capillary dysplasia can only be definitively diagnosed by tissue examination, it is possible that cases have been missed in children with complex congenital heart disease who died without such tissue having been obtained. This could influence the apparent association of alveolar capillary dysplasia with obstructive left heart disease and atrioventricular septal defect as described in the literature review.
The presence of congenital heart disease additionally complicates the correct diagnosis of alveolar capillary dysplasia as the superimposition of clinical symptoms results in a labile clinical presentation not explained by the heart defect alone. In a majority of cases in the literature, the diagnosis was established at autopsy after days or weeks of vain intensive care treatment even if several authors have described the feasibility of lung biopsy even in those critically ill patients.Reference Eulmesekian, Cutz, Parvez, Bohn and Adatia 21 , Reference Inwald, Brown, Gensini, Malone and Goldman 22 Some of these infants with alveolar capillary dysplasia and obstructive left heart disease underwent initially surgical repair and had a post-operative course characterised by crises of pulmonary arterial hypertension resistant to pulmonary vasodilatators.Reference Lane, Siwik, Preminger, Stork and Spector 7 , Reference Rabah and Poulik 8 , Reference Taborosi, Tödt-Pingel, Kayser and Dittrich 14 Only in few cases in the literature and in both infants presented here, diagnosis was established at lung biopsy.Reference Lane, Siwik, Preminger, Stork and Spector 7 Performance of lung biopsy contains the risk of clinical deterioration of the infant. On the other hand, the certitude of the fatal diagnosis makes it possible to withdraw the infant from costly and invasive treatment modalities once parental consent is obtained. Early diagnosis in patients with latent clinical presentations as in the case of our second patient can even permit a discharge from hospital if lung biopsy is well tolerated, allowing for a compassionate treatment at home.
In 2009, Stankiewicz et alReference Stankiewicz, Sen and Bhatt 17 identified for the first time a 16q24.1 microdeletion in four patients with alveolar capillary dysplasia and congenital heart disease. There are two other recent publications that have reported each another patient with a 16q24.1 microdeletion.Reference Yu, Shao, Kilbride and Zwick 18 , Reference Zufferey, Martinet and Osterheld 19 The deleted segment of chromosome band 16q24.1q24.2 includes the forkhead box gene cluster containing transcription factors FOXF1/FOXC2/FOXL1. Information concerning FOXF1 derives mainly from mouse models in which it participates in embryonic pulmonary, gastrointestinal, and endothelial development. Foxf1 haploinsufficiency in foxf1 −/+ mice is associated with lung hypoplasia and delayed lung maturation, leading to severe breathing problems and subsequently death within hours after birth in 50% of the newborn mice.Reference Mahlapuu, Enerbäck and Carlsson 23 FOXC2 is an important regulator of gene expression involved in angiogenesis and lymphatic vasculature. FOXC2 mutations are associated with lymphedema-distichiasis – double row of eyelashes – syndrome in the humans, which has also been reported in patients with tetralogy of Fallot, ventricular septal defect, and patent arterial duct.
Of the five patients with alveolar capillary dysplasia and heart defects reported by Stankievwicz et al, four had microdeletions encompassing FOXF1, FOXC2, and FOXL1. This concerned one infant with hypoplastic left heart syndrome, one with tetralogy of Fallot, and two patients with patent arterial duct. Only in one patient with partial atrioventricular septal defect a FOXF1 point mutation was identified. On the same line, the two other recently reported patients also harboured 16q24.1 microdeletions encompassing the fox gene cluster.Reference Yu, Shao, Kilbride and Zwick 18 , Reference Zufferey, Martinet and Osterheld 19 It has been hypothesised that alveolar capillary dysplasia results from haploinsufficiency of FOXF1 gene and that differences in the associated malformations are related to haploinsufficiency of other genes, namely, FOXC2 and FOXL1. Reference Stankiewicz, Sen and Bhatt 17
We identified for the first time a balanced reciprocal translocation between chromosomes 1 and 16 in the first infant of our series whose congenital heart defect was atrioventricular septal defect. The breakpoint on chromosome 16 lies ∼60 kb upstream of the FOXF1 gene. The translocation breakpoint junction may have exerted a position effect on the expression of the FOXF1 gene. A position effect is defined as a deleterious change in the level of gene expression brought by a change in the position of the gene relative to its normal chromosomal environment such as regulatory sequence elements. The possibility of a position effect exerted on the transcriptional regulation of this gene has already been suggested. Indeed, Stankiewicz et al also reported two patients with microdeletions located about 52 kb and 259 kb upstream of FOXF1 gene. Interestingly, these two patients had no heart defect and only one of them had other congenital malformations (imperforate anus).Reference Stankiewicz, Sen and Bhatt 17 Otherwise, cardiac malformations have only been described in patients with deletions encompassing the neighbouring FOXC2 and FOXL1 genes, suggesting that one of these genes may also contribute to the phenotype. In our patient, these two genes are present, but may have also been dysregulated secondary to a position effect.
Unfortunately, there was no genetic material available for FOXF1 analysis in the second infant of this report. Appropriate genetic studies in all patients with alveolar capillary dysplasia and congenital malformations including heart defects should be performed in order to deepen our understanding of the genetic association of FOX gene cluster defects in this rare congenital disorder.
Conclusion
In summary, diagnosis of alveolar capillary dysplasia associated with congenital heart disease should be considered in any newborn presenting with a particular instable clinical course not explained by usual haemodynamics and after exclusion of far more frequent reasons of concomitant neonatal respiratory distress such as sepsis, meconial aspiration, and asphyxia. Special attention should be paid if the infant presents a variant of left heart obstruction or atrioventricular septal defect associated or not with extracardiac congenital malformations. If in doubt, lung biopsy can be performed at the same time as cardiac repair or even in an instable infant on intensive care treatment. Early diagnosis can help to limit futile and aggressive treatments. Finally, detection of microdeletion or mutation of the FOXF1 gene should be considered in all patients who have alveolar capillary dysplasia with or without congenital heart disease, especially if other congenital malformations are present, as an accurate genetic diagnosis is important for genetic counselling.
Acknowledgements
We thank Dr Bettina Bessières and Unit of Foetopathologie, Department of Histo-embryology and Cytogenetics for fruitful comments on the histological and immunohistochemical results of the lung biopsies of the presented cases.






