


CHD is one of the most common birth anomalies, detected in 4–50 of every 1000 live births, and genetic factors have been suggested to underlie its development.Reference Hoffman and Kaplan 1 – Reference Huang, Liu, Sun, Lv, Du and Fan 3 Recent advances in genetics have led to the identification of several disease-causing genes in syndromic disorders associated with CHD.Reference Pierpont, Basson and Benson 4 Holt–Oram syndrome (HOS; OMIM142900) is a prototype of CHD and caused by mutations in the TBX5 gene. TBX5 is a transcriptional factor involved in heart and limb development.Reference Huang 5 – Reference Fan, Liu and Wang 7 Germline TBX5 mutations have been identified in 30–75% of Holt–Oram syndrome patients.Reference Adamopoulos, Kokkinou, Parissis and Kremastinos 8 – Reference Heinritz, Shou, Moschik and Froster 10
Holt–Oram syndrome is characterised by cardiac and upper- limb anomalies. It is a very rare condition, affecting 1 in 100,000 live births and inherited in an autosomal dominant manner.Reference Huang 5 , Reference Adamopoulos, Kokkinou, Parissis and Kremastinos 8 , Reference Heinritz, Shou, Moschik and Froster 10 – Reference Basson, Cowley and Solomon 12 Its phenotypic penetrance and expressivity have been known to vary among patients. However, upper-limb malformations are almost always present, and the preaxial radial ray distributed area including the radial, carpal, or thenar bones are mainly affected.Reference McDermott, Bressan and He 9 , Reference Basson, Cowley and Solomon 12
The prevalence of CHD in Holt–Oram syndrome patients has been reported to range from 75 to 95%, with atrial septal defect and ventricular septal defect being the most common CHDs in such patients. Some patients also develop cardiac rhythm disturbances such as sinus bradycardia or various degrees of atrioventricular conduction abnormalities.Reference Basson, Huang and Lin 6 , Reference Basson, Cowley and Solomon 12 – Reference Bossert, Walther, Gummert, Hubald, Kostelka and Mohr 14
In our current study, we describe the clinical characteristics of eight patients with Holt–Oram syndrome from six families focusing on cardiac manifestations. Germline TBX5 mutations were screened in all patients, and the SALL4 gene was analysed in patients without a TBX5 mutation to rule out Okihiro syndrome, which shows similar radial ray defects to Holt–Oram syndrome.Reference Kohlhase, Chitayat and Kotzot 15 Furthermore, given that TBX5 is involved in cardiac septal formation by interacting with other cardiac transcriptional genes, such as NKX2.5 or GATA4, we also investigated these two genes as potential new genetic causes of Holt–Oram syndrome.Reference Akazawa and Komuro 16 – Reference Maitra, Schluterman and Nichols 20
Patients and methods
A total of eight patients with Holt–Oram syndrome from six unrelated Korean families were included. We reviewed the clinical findings of each patient: demographic data, structural cardiac anomalies, electrocardiographic findings, skeletal anomalies, and surgical history. The clinical diagnosis of Holt–Oram syndrome was based on clinical or radiographic evidence of radial ray defect with associated cardiac anomalies. This study was approved by the Institutional Review Board of the Asan Medical Center, Seoul, Korea.
Molecular characterisation
Genetic analysis was performed using genomic DNA from peripheral blood using the Quick Gene Blood kit (Fujifilm, Tokyo, Japan). A total of nine exons of the TBX5 gene and their respective exon–intron boundaries were evaluated. The SALL4 gene was analysed in patients without a TBX5 mutation to rule out Okihiro syndrome. Two candidate genes, NKX2.5 and GATA4, were also evaluated. The primers of each gene were designed based on Primer3web (version 4.0.0; http://primer3.wi.mit.edu) of the Whitehead Institute: TBX5 (GenBank accession number: NT_009775.14.), NKX2.5 (NM_004387), GATA4 (NM_002052), and SALL4 (NT_011362.10) (Supplementary table S1). DNA sequencing was performed using the Big Dye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, California, United States of America). Multiple ligation-dependent probe amplification analysis was also performed for the detection of deletions and duplications in the TBX5, NKX2.5, GATA4, and SALL4 genes. A multiple ligation-dependent probe amplification probe mix (P180/P311, MRC, Amsterdam, The Netherlands) was used according to the manufacturer’s instructions.
Results
Patient characteristics
A total of eight patients, including three men and five women, were clinically diagnosed with Holt–Oram syndrome. Of the patients, four – patients 1, 2, 3, and 4 – had familial cases of Holt–Oram syndrome. In two patients, patients 5 and 6, Holt–Oram syndrome was suspected during the foetal period by abnormal foetal sonographic findings including ventricular septal defect and radial agenesis. Another four patients – patients 1, 3, 7, and 8 – were diagnosed postnatally because of cardiac and/or limb anomalies. Patient 2, father of patient 1, and patient 4, a daughter of patient 3, were identified by familial screening (Table 1). All patients had cardiac malformations: partial atrioventricular septal defect in one patient, combined ventricular septal defect and atrial septal defect in five patients, ventricular septal defect in one patient, and secundum atrial septal defect in one patient. The type of ventricular septal defect was perimembranous (four patients), muscular (one patient), or perimembranous combined with multiple muscular (one patient). The type of atrial septal defect was secundum in all patients. Combined limb anomalies were isolated thumb anomalies or various combined lesions including triphalangeal thumb (one patient), thumb hypoplasia (four patients), thumb aplasia (one patient), first to second digits syndactyly (one patient), complete radial aplasia (two patients), radial hypoplasia (one patient), metacarpal bone hypoplasia (one patient), or aplasia (one patient). These anomalies were bilateral in three patients (Fig 1).
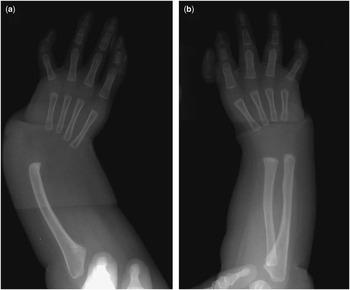
Figure 1 Plain X-ray findings for patient 7. ( a ) Radial aplasia, first metacarpal bone aplasia, and thumb hypoplasia in the left forearm. ( b ) Aplasia of the right first metacarpal bone.
Table 1 Clinical characteristics of the study patients.

ASD=atrial septal defect; CAVB=complete atriaoventricular block; Lt=left; PAB=pulmonary artery banding; pAVSD=partial atrioventricular septal defect; PDA=patent ductus arteriosus; PHT=pulmonary hypertension; pmVSD=perimembranous ventricular septal defect; PPM=permanent pacemaker; Rt=right; SVC=superior vena cava
Clinical outcomes of cardiac manifestations
The total follow-up period was 8.2±5.4 (range 2.7–14.9) years. Of the patients, seven underwent cardiac surgery at the average age of 4.0±4.7 (range 0.2–11.0) years. Early surgical intervention was required in three patients (patients 1, 5, and 8) owing to severe pulmonary hypertension accompanying ventricular septal defect and atrial septal defect. Of them, one patient (patient 5) underwent pulmonary artery banding as staged palliation owing to multiple apical muscular ventricular septal defects and poorly formed apical septum. The other patients underwent one-stage total correction and there were no residual shunt or significant valve regurgitation in these patients during follow-up.
In terms of arrhythmias, patient 3 showed sinus node dysfunction with severe sinus bradycardia after atrial septal defect operation. Initially it was mild and not symptomatic. However, after 10 years of operation, she intermittently experienced fatigue and dizziness. On routine regular health examination, severe bradycardia was detected and she was recommended to insert permanent pacemaker. It brought her to our hospital to further evaluation. Her treadmill exercise test showed no significant abnormality, and holter monitoring showed sinus bradycardia with intermittent premature atrial complex. Therefore, we decided to just observe and regularly follow-up. Her daughter (patient 4) also experienced arrhythmias after ventricular septal defect and atrial septal defect patch closure. In her case, complete atrioventricular block was developed postoperatively and required permanent pacemaker insertion. Complete atrioventricular block was noted in patient 8 postoperatively, which was thought as complication after large perimembranous ventricular septal defect patch closure operation. Preoperatively, she suffered from significant congestive heart failure requiring mechanical ventilation. She underwent total correction on day 67 with body weight of 3.5 kg at the operation. She performed permanent pacemaker insertion 24 days after total correction. Patient 7 also showed complete atrioventricular block after operation. During immediate postoperative periods, she had normal sinus rhythm with intermittent first-degree atrioventricular block. However, it progressed to 2:1 atrioventricular block with heart rate of 110 beats/minute at the time of discharge. On postoperative follow-up, it progressed to complete atrioventricular block that she underwent permanent pacemaker insertion on postoperative day 48.
Molecular characterisation of candidate genes for Holt–Oram syndrome
There were three different TBX5 mutations detected in three of the six families (50%): c.709C>T (p.Arg237Trp), c.880G>T (p.Glu294X), and c.755+2T>C (IVS7(+2)T>C). In the three patients with no TBX5 mutations, whole exons and their exon–intron boundaries of the SALL4 genes were investigated, but no mutations were found. Mutation analyses of NKX2.5 and GATA4 genes were negative (Table 2). Multiple ligation-dependent probe amplification detected no significant deletions or duplications in the TBX5, NKX2.5, GATA4, and SALL4 genes.
Table 2 Molecular characterisation of Holt–Oram syndrome candidate genes using direct sequencing and multiple ligation-dependent probe amplification.

* Normal, no exonic, and intron–exon boundary sequence variant, deletion, or duplication was found
Discussion
Holt–Oram syndrome is characterised by distinctive cardiac and upper-limb skeletal abnormalities. Although various cardiac anomalies such as isolated left atrial isomerism, inferior caval vein interruption with hemiazygous continuation to the left superior vena cava, and agenesis of pericardium and hypoplastic left heart syndrome have been reported in association with Holt–Oram syndrome. Holt–Oram syndrome most commonly presents as atrial septal defect or ventricular septal defect.Reference Huang 5 , Reference Bossert, Walther, Gummert, Hubald, Kostelka and Mohr 14 , Reference Manning, Kaufman and Roberts 21 In our present study, all eight patients showed cardiac septal defects. Various electrocardiographic abnormalities have also been reported in Holt–Oram syndrome patients with a wide range in incidence (18–71%). An abnormal electrocardiographic finding with no structural cardiac anomaly has been found in some patients (39%),Reference Newbury-Ecob, Leanage, Raeburn and Young 13 , Reference Bruneau, Logan and Davis 22 whereas others present with severe atrioventricular block requiring permanent pacemakers.Reference Huang 5 , Reference Bossert, Walther, Gummert, Hubald, Kostelka and Mohr 14 , Reference Holt and Oram 23 Half of the patients (four cases) experienced conduction abnormalities after cardiac surgery, and three of them ended up with performing permanent pacemaker insertion, which indicates the importance of careful evaluation and management of cardiac arrhythmias during follow-up.
The cardinal manifestations of limb anomalies accompanied by Holt–Oram syndrome are preaxial radial ray abnormalities without ulnar ray anomalies, as was noted in all of the patients in our present series. Some patients with SALL4-related disorders including Okihiro syndrome can also present with radial ray malformations. Townes–Brocks syndrome shares an overlapping phenotype with Holt–Oram syndrome, but the former condition is caused by SALL1 mutations. However, extra-cardiac or limb anomalies, including in the eye, kidney, and anus, are more common in SALL4-related disorders and Townes–Brocks syndrome than in Holt–Oram syndrome, which are important clues in the differential diagnosis.Reference Kohlhase, Chitayat and Kotzot 15 , Reference Powell and Michaelis 24 On the basis of these characteristics, all of our patients were diagnosed with Holt–Oram syndrome.
The mutation detection rate of the TBX5 gene in Holt–Oram syndrome patients is known to vary from 30 to 75%.Reference Borozdin, Bravo Ferrer Acosta and Bamshad 25 In the present analysis, the positive mutation rate was 50%. Of the three mutations, c.709C>T (p.Arg237Trp) and c.755+2T>C (IVS7(+2)T>C) are those previously reportedReference Basson, Huang and Lin 6 ,25, Reference Heinritz, Moschik and Kujat 26 whereas c880G>T (p.Glu294X) is the novel mutation that we detected in the family 1 (patients 1 and 2).Reference Lee, Kim, Kim, Kim and Yoo 27 Large exonic deletions in TBX5 have also been found in cases of Holt–Oram syndrome,Reference McDermott, Bressan and He 9 , Reference Borozdin, Bravo Ferrer Acosta and Bamshad 25 but such deletions were not detected in our patients.
The variable mutation detection rate of the TBX5 gene among studies may be explained by the wide spectrum of clinical manifestations among patients with Holt–Oram syndrome. It can be argued that applying strict diagnostic criteria, including personal and/or family history of cardiac septal defect and/or conduction defects in the presence of preaxial radial ray deformity, can improve the detection rate of genetic causes of Holt–Oram syndrome.Reference McDermott, Bressan and He 9 However, in this study, all patients fulfilled strict diagnostic criteria, but the mutation detection rate was only 50%.
There is much controversy regarding the genotype–phenotype correlation of Holt-Oram syndrome. Basson et alReference Basson, Huang and Lin 6 suggested that the location of mutations in the TBX5 gene has an impact on the type of anomaly, that is, cardiac or limb, but this correlation was opposed by other groups. Owing to the small number of patients included in our study, evaluation of genotype–phenotype correlations was limited. Considering the three mutations identified in our study, c.880G>T (p.Glu294X), c.755+2T>C (IVS7(+2)T>C), and c.709C>T (p.Arg237Trp), previous studies showed that most patients with p.Arg237Trp have limb anomalies, whereas only half have cardiac anomalies;Reference Basson, Huang and Lin 6 in contrast, our patient harbouring p.Arg237Trp had both cardiac and limb anomalies. In addition, we were unable to identify any significant differences in phenotypic expressivity between patients with and without TBX5 mutations.
Discovering genetic factors underlying cardiac manifestations in Holt–Oram syndrome is important not only to better understand the molecular pathology of cardiac lesion but also to help families affected by Holt–Oram syndrome, by providing them with genetically appropriate counselling and care. Genetic screening is an effective tool to identify affected family members who have no obvious manifestations of Holt–Oram syndrome. Moreover, owing to the dominant inheritance pattern of Holt–Oram syndrome, prenatal genetic counselling, based on identification of familial mutations, can be an important issue in preparing perinatal care and family planning. Therefore, further efforts are needed to identify new causative genes or genetic influences.
To increase the mutation detection rate, we screened for SALL4 point and exonic deletion mutations, which are found in Okihiro syndrome as well as in rare cases of Holt–Oram syndrome.Reference Kohlhase, Chitayat and Kotzot 15 , Reference Kohlhase, Schubert and Liebers 28 However, no SALL4 mutations were identified. The interaction of TBX5 with other transcription factors such as NKX2.5, GATA4, TBX20, TBX1, and MYH6 may affect cardiac embryogenesis in Holt–Oram syndrome patients.Reference Granados-Riveron, Pope and Bu’lock 19 , Reference Bruneau 29 – Reference Postma, van de Meerakker and Mathijssen 31 Screening for interacting candidate genes may help to identify new causative genes of cardiac manifestations in Holt–Oram syndrome. We therefore screened NKX2.5 and GATA4, which were known to interact with TBX5 during cardiac septal formation, myocardial patterning, and chamber specification, in an attempt to identify new disease-causing genes in Holt–Oram syndrome, but found no positive results. Furthermore, multiple ligation-dependent probe amplification analysis did not reveal any exonic deletion or duplication in these candidate genes. It might be because of the small number of affected patients in this study.
Conclusions
Most commonly detected cardiac anomalies in Holt–Oram syndrome were cardiac septal defects. Our present data indicate the importance of careful management and monitoring for CHDs in patients with Holt–Oram syndrome, especially in those with cardiac conduction abnormalities. Although our patients showed typical clinical findings and underwent TBX5 and other candidate gene screening, half of all Holt–Oram syndrome patients remain genetically undiagnosed. Further large-scale genomic research is needed to uncover new genetic causes and genotype correlation with cardiac manifestations in patients with Holt–Oram syndrome and understand the genetic heterogeneity of this disease.
Acknowledgements
None.
Financial Support
This research was supported by a grant from the National Research Foundation of Korea, funded by the Ministry of Education, Science and Technology (Grant No. 2011-0019674 and 2012R1A1A1041513).
Conflicts of Interest
None.





